
Deep learning has become the default answer to almost every hard problem in computational biology, but it is not always the right one. Applying deep learning to biology research pays off when raw, high-dimensional inputs such as images, sequences, or interaction networks carry more signal than any hand-engineered feature set can capture. This guide walks through what deep neural network architectures add over classical machine learning (ML), and where the added complexity is not worth the cost.
Key takeaways
- Deep learning earns its complexity when data are unstructured, such as images, sequence data, or network data, and loses that advantage on small, well-annotated tabular datasets.
- Convolutional neural networks (CNNs) remain the standard architecture for biological imaging tasks such as cell segmentation and image restoration.
- Transformers, the architecture behind large language models, now dominate sequence-based genomics and protein modeling because of their ability to capture long-range dependencies.
- Graph neural networks (GNNs) extend deep learning to network-structured biological data such as protein interaction maps and metabolic pathways.
- Starting with deep learning no longer requires a dedicated graphics processing unit (GPU) cluster; pretrained models and cloud-based notebook environments have substantially lowered the entry barrier.
Deep learning vs classical ML in biology research
Deep learning’s core advantage over classical ML is representation learning: instead of relying on features a researcher engineers by hand, a neural network learns which patterns in raw data matter for the task at hand. This distinction, one piece of the wider AI and data science toolkit available to life scientists, determines when the additional complexity of a neural network is worth adopting for biological data analysis.
Classical ML methods, particularly tree-based ensembles such as random forests and gradient boosting, remain the stronger choice for structured tabular data, including clinical variables, quality control metrics, and engineered genomic features. These tree-based ensemble methods handle mixed data types natively, resist overfitting with minimal tuning, and produce interpretable feature importance scores that connect a prediction back to specific biological variables.
Neural networks earn their added complexity when the data are unstructured or when the underlying biological signal is distributed across raw sequence data, pixel data, or network structure in ways that manual feature engineering cannot fully capture. Recognizing which category a dataset falls into is the single most useful skill for deciding whether to reach for deep learning tools in bioinformatics at all, and it is a decision that should always precede any discussion of specific architectures.
The trade-off runs in both directions. A neural network trained on a small, well-annotated tabular dataset typically offers no accuracy advantage over a tuned tree-based model, and it introduces training instability, longer development time, and reduced interpretability that a biology research team may not be equipped to manage. Conversely, applying a tree-based model to raw pixels or unaligned sequence data forces a researcher to hand-engineer features that a neural network would otherwise learn automatically, often at a real cost to accuracy.
CNN biology applications for imaging and sequence data
CNNs remain the workhorse architecture for extracting spatial patterns from biological images, and CNN biology applications now span cell segmentation, super-resolution restoration, and high-content phenotypic screening. Convolutions detect local patterns, such as edges or textures in a fluorescence image, and stack those local detections into progressively more abstract representations of cellular structure.
Cell segmentation illustrates this advantage clearly. Generalist CNN-based segmentation tools can now delineate cells and nuclei across a wide range of imaging modalities without retraining for each new experimental context, a substantial improvement over the manual and specialist methods that preceded them, as demonstrated in the widely adopted generalist cell segmentation algorithm introduced by Stringer and colleagues.
CNNs also underpin many sequence-based genomics tools that treat DNA as a one-dimensional signal, scanning fixed-length windows to detect motifs associated with regulatory activity. This approach works well for local sequence patterns, but the fixed, local receptive field of a convolution limits how far a CNN can look for distant regulatory elements, a constraint that becomes important when comparing convolutional and attention-based architectures for genomic sequence tasks.
Training a CNN from scratch still requires a reasonably large, well-labeled image dataset, which is not always available for a niche experimental system or an unusual imaging modality. In practice, most biology research teams start from a generalist pretrained CNN and fine-tune it on a smaller set of images specific to their own experiment, which substantially reduces both the data and compute required compared with training an entirely new architecture.
RNNs and transformers for life sciences sequence data
Recurrent neural networks (RNNs) were the first deep learning architecture built to process biological sequences step by step, carrying forward a memory of earlier positions as they moved along a strand of DNA, RNA, or protein. This sequential design made RNNs a natural fit for variable-length biological sequences, but training them is slow and they struggle to retain information across very long distances.
Transformers solved this long-range problem by replacing sequential processing with self-attention, allowing every position in a sequence to directly weigh the relevance of every other position regardless of distance. This shift has made transformers the dominant architecture for genomics and protein-modeling applications in the life sciences. Enformer, a transformer-based model for predicting gene expression directly from DNA sequence, integrates regulatory information from elements up to 100,000 base pairs away, a receptive field five times larger than the CNN-based models it replaced, as detailed in its original architecture paper.
Protein language models extend the same transformer architecture to amino acid sequences. These models learn evolutionary and structural patterns from massive protein sequence databases without explicit structural labels, and the largest versions have demonstrated that atomic-level structural information emerges directly from sequence alone as the models scale, a finding reported in the evolutionary-scale protein language model study that introduced ESMFold. Researchers evaluating neural networks and transformers for their own sequence problems should weigh this scaling advantage against the substantial computational cost of training or fine-tuning models at this size.
RNNs are still used in settings where sequence length is short and a full transformer’s computational overhead is not justified, and hybrid architectures that combine convolutional feature extraction with recurrent or attention layers remain common in specialized genomics tools. The practical takeaway for most researchers is that transformers have become the default choice for new sequence-modeling projects in biology research, while RNNs persist mainly in legacy tools and computationally constrained settings.
Graph neural networks for molecular and network biology
GNNs extend deep learning to data that is naturally structured as a network of relationships rather than a sequence or a grid of pixels. Molecules, protein-protein interaction maps, and metabolic pathways are all more accurately represented as graphs, with atoms or proteins as nodes and their interactions as edges, than as any fixed-length sequence or table.
A GNN learns node representations by repeatedly aggregating information from each node’s neighbors, allowing the model to capture both a node’s individual properties and the structural context in which it sits. This makes GNNs well suited to predicting protein function from interaction context or modeling how a genetic perturbation propagates through a regulatory network, tasks where a molecule’s biological role often depends as much on what it connects to as on its own properties. A study applying GNNs to protein-protein interaction prediction, combining protein structural and sequence features, found that its graph-based approach outperformed most prior methods that relied on sequence information alone.
GNNs depend heavily on the quality of the underlying graph structure, and a poorly defined or incomplete interaction network will limit model performance regardless of architecture choice. They also require more careful implementation than CNNs or transformers, since standard deep learning libraries offer less mature, off-the-shelf support for graph-structured biological data, which raises the practical entry cost for a research group without prior GNN experience.
Table 1: How CNNs, transformers, and GNNs map onto common biological data types and tasks.
|
Architecture |
Best-suited data type |
Representative biological task |
|
CNN |
Images and short, local sequence windows |
Cell and nucleus segmentation, motif detection |
|
Transformer |
Long sequences with distant dependencies |
Gene expression prediction, protein language modeling |
|
GNN |
Network-structured relational data |
Protein interaction prediction, pathway modeling |
When deep learning outperforms classical ML in biology research
Deep learning is genuinely better than classical ML when a problem’s biological signal cannot be reduced to a manageable set of hand-engineered features without losing information that matters for the prediction. Protein structure prediction is the clearest example: decades of physics-based and homology-based methods fell far short of experimental accuracy until a deep learning system trained directly on sequence and structural data closed that gap almost entirely, as demonstrated in the landmark AlphaFold structure prediction paper.
The same pattern holds across genomics and imaging: whenever the relevant biological signal is distributed across raw pixels, long genomic sequences, or complex interaction networks, deep learning architectures that learn directly from that raw structure outperform methods that require the signal to be pre-summarized into a fixed feature set. The complexity is not worth it, however, when a dataset is small, well-annotated, and already reducible to a modest number of informative tabular features, since a well-tuned classical model will often match a poorly tuned neural network at a fraction of the computational cost and with far greater interpretability.
A simple framework helps make this decision systematic rather than reflexive:
- Identify whether the raw data are images, long sequences, or network-structured relationships, or whether they are already reducible to a modest set of tabular features.
- Check whether enough labeled or pretrainable data exist to support a deep model, since neural networks generally need far more examples than tree-based methods to avoid overfitting.
- Weigh interpretability requirements against predictive ceiling, since deep models are typically harder to explain than tree-based models, which produce interpretable feature importance scores.
- Establish a classical ML baseline before investing in a deep learning pipeline, so the added complexity can be justified by a measurable performance gain.
- Revalidate any deep learning model on data that reflects the real deployment context, including new platforms, tissues, or populations.
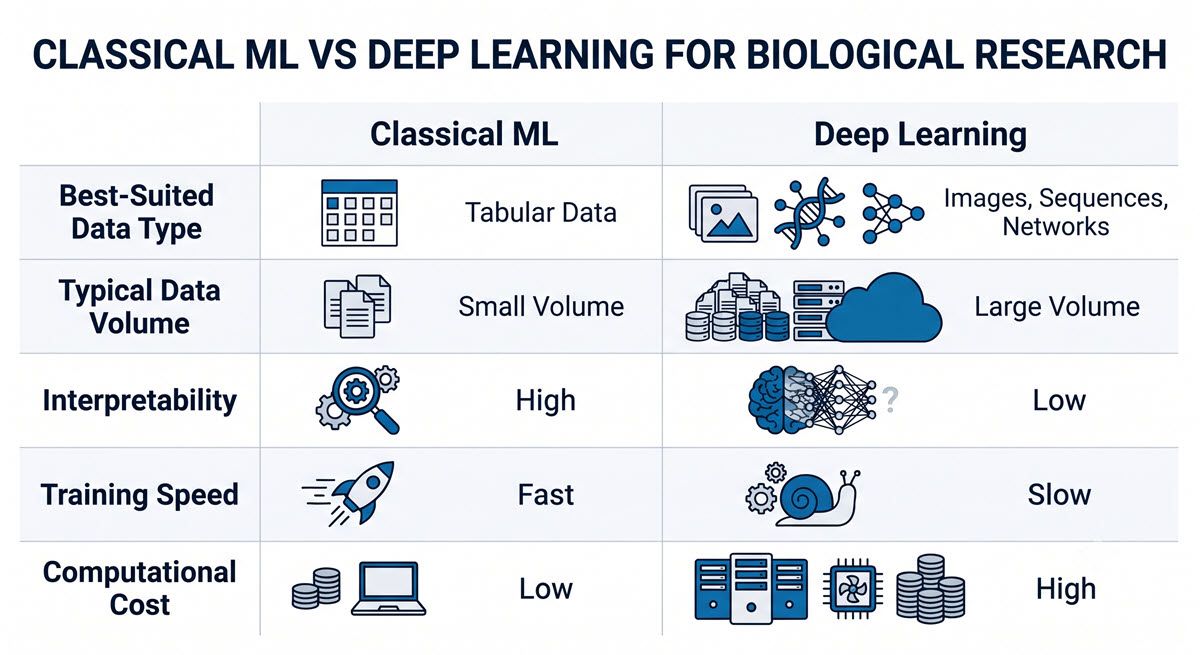
Figure 1: A side-by-side comparison of classical ML and deep learning across data type, data volume, interpretability, training speed, and computational cost. Credit: AI-generated image created using Google Gemini (2026).
How to start deep learning research without a GPU cluster
Getting started with deep learning in bioinformatics no longer requires access to an institutional GPU cluster or other dedicated supercomputing hardware. Pretrained models for CNNs, transformers, and GNNs are now widely distributed through open-source repositories, and many can be fine-tuned or applied directly to new data using free or low-cost cloud-based notebook environments rather than local hardware.
Funding agencies have also lowered this barrier directly. NIH-supported researchers can access discounted commercial cloud computing, including machine learning and artificial intelligence infrastructure, through the NIH cloud computing initiative, which removes the need for a lab to maintain its own high-performance computing hardware. Starting with a pretrained model and fine-tuning it on a smaller, task-specific dataset is almost always more practical than training a deep architecture from scratch, and it is the approach used by most working computational biologists outside dedicated machine learning groups.
A practical starting sequence looks similar across CNNs, transformers, and GNNs: identify a pretrained model built for a closely related biological task, evaluate its out-of-the-box performance on a small labeled subset of the target data, and only invest in fine-tuning or architecture changes once that baseline reveals a specific, measurable gap. This sequence keeps compute costs modest and gives a research group an early, honest signal about whether deep learning tools for bioinformatics are actually solving the problem at hand before committing further time or budget.
Matching deep learning to biology research problems
Deep learning for biology research delivers real, measurable gains when the underlying data are images, long sequences, or interaction networks that resist reduction to a fixed set of engineered features. CNNs, transformers, and GNNs each map onto a specific data structure, and matching the architecture to that structure, rather than defaulting to whichever approach is most discussed, is the decision that determines whether a project succeeds.
For structured, tabular biological data with a modest number of well-annotated examples, classical tree-based methods remain the more practical starting point, and the added complexity of a neural network should be justified by a measured performance gain over that baseline. This decision framework applies across the broader landscape of machine learning methods for life science research, of which deep learning is one powerful but situational tool among several.
This content includes text that has been created with the assistance of generative AI and has undergone editorial review before publishing. Technology Networks’ AI policy can be found here.

