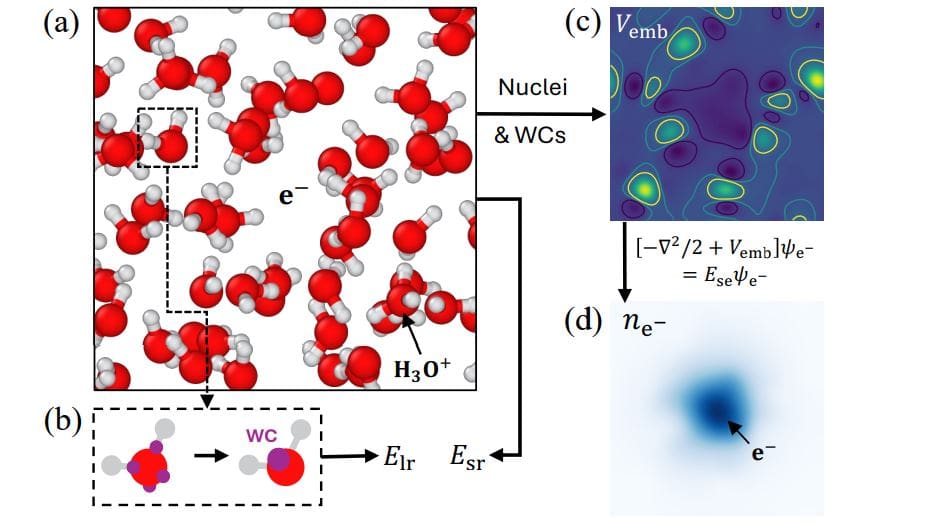
Understanding the behavior of electrons in liquids is a major challenge for computational chemistry, as traditional methods have difficulty modeling these particles accurately without incurring excessive computational costs. Ruiqi Gao of Princeton University, Pinchen Xie of Lawrence Berkeley National Laboratory, Roberto Car of Princeton University and colleagues have developed a new machine learning approach that incorporates the mechanical behavior of excess electrons directly into molecular simulations. This innovative method achieves accuracy comparable to complex quantum calculations, but at a fraction of the computational cost, and allows researchers to study the interactions of solvated electrons in detail. The research team successfully applied this technique to model the reaction of electrons and hydronium ions in water, identifying the specific proton transfer mechanism and accurately predicting reaction rates and free energies in close agreement with experimental observations, providing new insights into fundamental chemical processes.
Density functional theory calculations provide a computationally efficient approach to achieving accurate results, but electrons are difficult to model explicitly. Scientists have developed an electron-enabled machine learning force field that addresses this limitation by mechanically handling excess electron quanta while using machine learning to reproduce density functional theory calculations of the remaining interactions. This method accurately models the behavior of electrons in water and their reactions with hydronium ions, revealing the proton transfer mechanism by which protons recombine with electrons. The research team determined forward reaction rates between 350 K and 450 K and demonstrated an Arrhenius relationship between activation energy that is consistent with existing experimental measurements.
QM/ML force field development and validation
This study details the methodology used to investigate the reaction between electrons and hydronium ions to form neutral hydrogen atoms. This research focuses on utilizing a combined quantum mechanics/machine learning (QM/ML) force field to accurately calculate reaction rates and determine equilibrium constants. This innovative approach combines the precision of quantum mechanical calculations with the efficiency of machine learning to address the challenge of accurately simulating this reaction. The authors carefully validated their method and demonstrated the reliability of their results.
This study performed detailed calculations of response rates using survival probability methods and highlighted the importance of sampling from a quasi-stationary distribution to avoid biased estimates. A finite size scaling analysis was performed to extrapolate the rate constants to the dilution limit to ensure accuracy. Equilibrium constants were calculated using mean force potential calculations and free energy integration, and were enriched with collective variables to improve sampling efficiency. Convergence of the mean force potential with increasing system size was demonstrated, confirming the robustness of the calculations.
The research team investigated the energy and spatial extent of the excess electrons and correlated the modeled results with Kohn-Sham calculations from density functional theory. The difference between the modeled and DFT results is due to the absence of self-interactions in the model, highlighting room for future improvement. A detailed analysis of the log survival probability method was provided, showing the results obtained over time. The authors critically evaluated the limits of the potential of standard neural networks for this response, explaining its instability and inability to accurately capture its dynamics. They argue for the need for more physical processing of excess electrons and justify the QM/ML approach. The development of QM/ML force fields, the use of collective variables, finite size scaling analysis, and addressing self-interaction errors represent important innovations.
Confirming the reaction mechanism of hydronium ions using a model
Scientists have developed a new way to model the behavior of electrons in water and their reactions with hydronium ions, achieving both accuracy and efficiency through a physics-based approach. The simulations revealed the proton transfer mechanism governing the recombination of excess electrons and protons and determined the reaction rates and equilibrium constants over different temperature ranges. The calculated activation energies are consistent with existing experimental measurements, confirming the accuracy of the model. Comparisons with previous studies at 400 K further validated the results. The research team also determined the equilibrium constant for the reaction, allowing calculation of the reaction free energy, which also agrees with experimental data.
Enhanced sampling simulations were used to map the mean force potential, which is important for determining the equilibrium constant. The results showed a van’t Hoff plot of pK1 decreasing with increasing temperature, indicating an endothermic response and reflecting the experimental trend. The reaction free energy calculated at 350 K is in close agreement with the experimental value. Diffusion coefficients for water, solvated electrons, and hydronium ions were measured, and values were obtained that showed reasonable agreement with established data. This study highlights the dramatic stabilizing effect of the solvent environment, as reactions in vacuum require much more energy than observed in water. The developed model is extensible and applicable to a wide range of solvated electron reactions, potentially advancing our understanding of nonadiabatic electron transfer processes and bound excitons.
Accurately model electron transfer in water
Researchers have developed a new machine learning method to model the behavior of electrons in water and their subsequent reaction with hydronium ions. This approach provides a mechanically accurate representation of the excess electrons while at the same time reproducing calculations derived from density functional theory. Through simulations, the researchers identified the proton transfer mechanism that governs the recombination of excess electrons with protons, determined reaction rates and equilibrium constants over different temperature ranges, and achieved results consistent with experimental data. The developed model has demonstrated accuracy, robustness, efficiency, and is designed to be easily extended to other systems. The researchers acknowledge the limitations of the underlying electronic structure methods and the neglect of nuclear quantum effects, and suggest that these areas are priorities for future research. They also expect the applicability of their method to a wide range of reactions involving solvated electrons, including nonadiabatic electron transfer, small polarons, and bound excitons, paving the way to further study these complex phenomena.