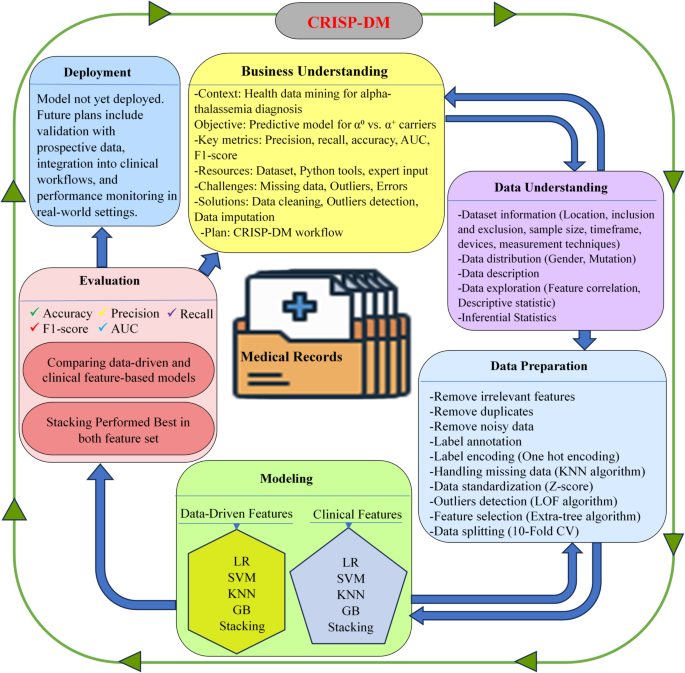
This study followed the Cross Industry Standard Process for Data Mining (CRISP-DM) as the main framework for project development. CRISP-DM is a well-structured methodology that has been widely used since the 1990s in both Knowledge Discovery in Databases (KDD) and modern data science projects23. Its popularity comes from its simplicity, repeatability, and effectiveness in guiding data mining projects24. CRISP-DM is also valued for being intuitive, adaptable, and closely aligned with standard data science workflows23. By ensuring a clear connection between project goals and technical implementation while allowing iterative improvements, it was an ideal choice for this study. The project followed the six CRISP-DM phases: business understanding, data understanding, data preparation, modeling, evaluation, and deployment25 as shown in Fig. 1.

Workflow Framework for the Detection of Common Large Alpha-Thalassemia Mutations Using Machine Learning.
Business understanding
This project was developed within the context of health data mining to improve diagnostic support for alpha-thalassemia carriers by analyzing patient laboratory data. The main goal was to develop a predictive model that would classify patients as carriers of either α⁰ or α⁺ thalassemia using routine hematological indices. The technical objective was to create a model with high predictive performance, evaluated by metrics such as precision, recall, and overall accuracy. During the initial phase, available resources including the dataset, computational tools (Python and associated libraries), alongside consultation with a genetics expert, were assessed. Key challenges, such as missing data, outliers, and noise were identified, with planned solutions like data cleaning, outlier detection and data imputation techniques. A structured CRISP-DM workflow diagram (Fig. 1) was created to outline the methodology and serve as a step-by-step execution plan.
Data understanding
This phase involves data collection, examination, and exploration to detect quality concerns, meaningful patterns.
Data collection
This retrospective study was approved by the Ethics Committee of Ahvaz Jundishapur University of Medical Sciences (AJUMS) under approval number IR.AJUMS.REC.1402.311, and the requirement for informed consent was waived in accordance with national regulations due to the anonymized nature of the data. All records were de-identified, and strict confidentiality and data protection standards were maintained throughout the study.
All methods were performed in accordance with the relevant guidelines and regulations stipulated by the Ethics Committee of Ahvaz Jundishapur University of Medical Science (AJUMS) and local data protection laws.
The data were collected from the Thalassemia and Hemophilia Research Center and Genetics Laboratory located at Dastgheib Educational and Medical Center in Shiraz. The center’s services include chorionic villus sampling (CVS), thalassemia prevention testing, fetal screening, coagulation factors and inhibitors testing, male infertility testing, and amniocentesis.
Inclusion criteria required patients to have documented common large deletions in the alpha-globin gene, including heterozygous α 3.7, heterozygous α4.2, homozygous α4.2, large deletions such as − α20.5 and −−Med, or compound heterozygous mutations (α 3.7/ α4.2). Additionally, patients had to have HbA2 levels below 3.5%. Exclusion criteria included cases where marriage counseling was discontinued, records that were corrupted or illegible, incomplete data due to sample depletion, follow-up loss resulting in missing or incomplete records and cases with coexisting iron deficiency.
The dataset included 956 patients, consisting of 435 females and 521 males (Fig. 2a). Of these, 506 were diagnosed with α⁰ thalassemia (two-gene deletions) and 450 with α⁺ thalassemia (single-gene deletion) (Fig. 2b). The α⁺ group comprised 234 cases of heterozygous α 3.7, 216 cases of heterozygous − α⁴.², 195 cases of compound heterozygous − α³.⁷/−α⁴.², and 38 cases of homozygous − α⁴.² mutations. The α⁰ group included 73 cases of − α²⁰.⁵ and 200 cases of −−Med deletions (Fig. 2c).

Data distribution in alpha-thalassemia dataset.
This research was conducted over a period spanning from 2001 to 2023. The study focused on individuals aged 15 to 45 diagnosed with alpha-thalassemia and referred for premarital hematological and genetic screening related to thalassemia. From a larger archive of 20,399 records on hemoglobinopathies, a subset of 1,167 cases with relevant alpha-thalassemia mutations was extracted for analysis. Following screening by a specialist to remove records with co-existing iron deficiency anemia and cases with significant missing data, a total of 956 eligible records were finalized.
Data were obtained from hospital records, including referral forms from health centers, complete blood count (CBC) results generated using a Sysmex KX-21 hematology analyzer, and genetic test reports. Genetic analyses for common point deletion-type mutations in the alpha- and beta-globin genes were performed using ARMS-PCR, while reverse dot blot techniques were employed in older records. Common large deletions associated with the alpha-globin gene were analyzed using GAP-PCR. Capillary electrophoresis was performed using Sebia and Helena V8 E-class devices, whereas older records were analyzed using cellulose acetate electrophoresis and high-performance liquid chromatography (HPLC). For complex cases, Sanger sequencing was performed using the Applied Biosystems 3130 XL Genetic Analyzer, and advanced deletion detection was carried out using Multiplex Ligation-dependent Probe Amplification (MLPA).
Data description
The dataset contained a total of 20 features extracted from the medical records of patients with alpha-thalassemia. Of these, 16 features were related to hematological indices derived from the evaluation of RBCs, white blood cells, and platelets, as measured by CBC analysis. An additional 3 features represented the levels of Hb fractions in the blood, obtained through Hb electrophoresis. Finally, genetic testing was used to identify the type of large deletion mutation present in alpha-thalassemia patients. The details of these features are presented in Table 1.
The distribution of hematological parameters used in this study is demonstrated in Fig. 3. The histograms provide an overview of how each parameter is distributed across the dataset, displaying the variability and range of values for RBC indices, WBC counts, PLT indices, and Hb fractions.

Histograms of hematological parameters in alpha-thalassemia patients.
Data exploration
The correlation matrix (Fig. 4) shows several notable relationships. First, Hb and RBC show a moderate positive correlation (0.48), which suggests that an increase in RBC count generally leads to higher Hb levels. In thalassemia, the defective Hb synthesis leads to a reduction in Hb content per RBC which consequently cause the body to compensate for reduced oxygen-carrying capacity by increasing RBC production. However, the correlation is not perfect because these RBCs are microcytic (small size) and hypochromic (pale), meaning they contain less Hb per cell. This results in a moderate positive correlation rather than a strong one. The negative correlation between RBC and MCV (-0.45) aligns with the pathophysiology of α-thalassemia, where increased RBC production compensates for ineffective erythropoiesis, but the newly produced RBCs are smaller in size, leading to lower MCV. Similarly, RBC shows a negative correlation with MCH (-0.39), indicating that as RBC count rises, the Hb content per cell declines, consistent with the microcytic and hypochromic nature of α-thalassemia. However, the weak negative correlation between RBC and MCHC (-0.2) suggests that despite a lower Hb content per cell, the relative concentration of Hb in the smaller RBCs remains somewhat stable, potentially due to a compensatory effect in Hb packing density. The relationships of Hb with MCV (0.48), MCH (0.6), and MCHC (0.62) further highlight these trends. Hb is positively correlated with MCV because larger RBCs typically contain more Hb. The stronger correlation between Hb and MCH (0.6) compared to Hb and MCV suggests that overall Hb content per cell is a more direct determinant of total Hb than cell size alone. The highest correlation, Hb with MCHC (0.62), suggests that the intracellular Hb concentration plays a significant role in determining total Hb levels, despite variations in RBC count and size. The strong correlation between MCV and MCH (0.9) reinforces the concept that larger RBCs tend to contain more Hb, as expected in normal erythropoiesis. The correlation between MCH and MCHC (0.82) further confirms that Hb content per RBC and its concentration within the cell are closely linked. Consequently, the moderate correlation between MCV and MCHC (0.55) indicates that larger RBCs tend to maintain a higher intracellular Hb concentration, though this relationship is not as strong as the MCV_MCH correlation due to variations in Hb synthesis efficiency. By integrating these relationships, the correlation between HCT and RBC (0.72) can be justified. HCT is calculated as [(RBC×MCV)/10]26, meaning that while MCV is reduced in α-thalassemia, the substantial increase in RBC offsets this effect, leading to an overall positive correlation with HCT. The very strong correlation between HCT and Hb (0.89) reflects that total Hb levels and HCT are directly related, both being dependent on RBC count and Hb content per RBC. Examining the RDW, its negative correlation with Hb (-0.31) suggests that greater variability in RBC size (anisocytosis) is associated with lower total Hb. In α-thalassemia, impaired Hb synthesis results in a heterogeneous population of RBCs with varying sizes and Hb content. The body compensates by increasing erythropoiesis, but many newly produced RBCs remain small and hypochromic, contributing to lower total Hb. The weak positive correlation between RDW and RBC (0.36) indicates that increased RBC count is associated with a wider distribution of cell sizes, likely due to the presence of both microcytic and relatively normocytic cells. The strong negative correlations of RDW with MCV (-0.66), MCH (-0.64), and MCHC (-0.46) highlight that greater size variation among RBCs is predominantly due to a higher proportion of smaller, hypochromic cells, reinforcing the hallmark microcytic hypochromic profile of α-thalassemia.

Feature Correlation Matrix of Key Hematological Parameters.
The usual proportion of Hb types in normal adults is 95% to 98% HbA, 2% to 3% HbA2, and < 2% HbF29, and the total sums to 100%. Given that HbA levels are largely stable in α-thalassemia carriers (97.45% in α⁺ and 97.31% in α⁰), and since HbA is the dominant fraction (~ 97%), even minor fluctuations in HbA2 and HbF can create noticeable inverse correlations with HbA. The negative correlation between HbA and HbF (-0.7) is justified by the observation that HbF is slightly higher in α⁰ compared to α⁺, suggesting that when HbA slightly decreases, HbF increases to maintain balance. Similarly, the negative correlation between HbA and HbA2 (-0.56) arises because HbA2 is slightly lower in α⁰ than in α⁺. In contrast, the correlation between HbF and HbA2 (-0.27) is weak, suggesting that their variations are largely independent.
Although the mean values for PLT count, MPV, PDW, and P-LCR remain within normal limits in both α⁺ and α⁰ thalassemia, a weak inverse correlation is observed between PLT and MPV (-0.31), PDW (-0.34), and P-LCR (-0.34). This suggests that individuals with relatively higher PLT counts tend to have slightly smaller and less heterogeneous platelets, while those with mildly lower PLT counts exhibit modest increases in MPV, PDW, and P-LCR. However, because these indices lie within the normal range and the correlations are weak, the relationship does not indicate a clinically significant trend. Meanwhile, the strong positive correlations between PDW and MPV (0.8), PDW and P-LCR (0.88), and MPV and P-LCR (0.9) suggest that as PLT size (MPV) increases, both the variation in PLT size (PDW) and the proportion of large platelets (P-LCR) also rise. This is expected, as larger platelets are typically younger and more reactive, contributing to greater size variability and a higher proportion of large platelets in circulation.
WBC subtypes are reported as percentages of the total white cell count, and under normal conditions, these percentages sum to approximately 100%. Typical reference ranges include 50–70% neutrophils, 1–5% eosinophils, 0–1% basophils, 2–10% monocytes, and 20–45% lymphocytes30. In this context, the strong negative correlation between Lym% and Nuet% (-0.95) indicates that when neutrophils rise, lymphocytes proportionally decrease. Meanwhile, the moderate negative correlation between the MXD% (which includes monocytes, eosinophils, and basophils) and Nuet% (-0.57) follows a similar pattern, although its effect is less pronounced because these cell types collectively account for a smaller fraction of total white blood cells. Ultimately, because these subpopulations must sum to 100%, any increase in one subset inevitably reduces the relative proportions of the others.
The correlation matrix also revealed a strong association between gender and key RBC parameters, with correlations of 0.6 for RBC count, 0.67 for Hb, and 0.72 for HCT. These differences align with known biological variations, where males typically exhibit higher Hb and HCT levels than females. This disparity is influenced by higher testosterone levels in men, which promote erythropoiesis, as well as genetic differences in the erythropoietin gene and its receptor between sexes31.
Additionally, Fig. 5, a network graph based on the correlation matrix visualizes relationships between variables. Each node represents a hematological parameter, while the connecting lines indicate correlation strength and direction. Thicker lines show stronger correlations, with red representing negative correlations and blue indicating positive ones.

Correlation Network Graph of Blood Parameters in Alpha-Thalassemia Carriers Datasets.
The descriptive statistics for both α⁺ and α⁰ thalassemia carriers are summarized in Table 2. Overall, distinct trends were observed between the two groups across various hematological and Hb fractions. In terms of RBC count, both groups showed elevated levels characteristic of α-thalassemia carriers, with α⁰ carriers exhibiting a higher mean RBC count (6.14 ± 0.59 × 10⁶/µL) compared to α⁺ carriers (5.69 ± 0.46 × 10⁶/µL). This reflects the compensatory erythropoiesis common in α⁰ thalassemia, where defective Hb synthesis leads to increased RBC production. The Hb levels were lower in α⁰ carriers (12.96 ± 1.34 g/dL) compared to α⁺ carriers (14.15 ± 1.44 g/dL), matching the more severe anemia in α⁰ thalassemia. Similarly, HCT levels followed this trend, being slightly lower in α⁰ carriers (42.10 ± 3.97%) than in α⁺ carriers (42.98 ± 3.56%). Despite higher RBC counts, this reduction is likely driven by the smaller size of RBCs in α⁰ individuals. The MCV and MCH values were notably reduced in α⁰ carriers (68.61 ± 3.29 fL and 21.12 ± 1.28 pg, respectively) compared to α⁺ carriers (75.56 ± 3.99 fL and 24.92 ± 1.86 pg, respectively), reflecting the more pronounced microcytic and hypochromic features of α⁰ thalassemia. MCHC was also slightly lower in α⁰ (30.77 ± 1.08 g/dL) than in α⁺ (32.89 ± 1.31 g/dL), indicating reduced Hb concentration per unit of red cell volume. RDW was elevated in α⁰ (15.14 ± 1.16%) relative to α⁺ (13.94 ± 1.01%), consistent with increased anisocytosis, a hallmark of α⁰ thalassemia.
For Hb fractions, HbA showed similar means in both groups (~ 97%), though slightly lower in α⁰ (97.31%) than α⁺ (97.45%). HbA2 levels were slightly reduced in α⁰ (2.30 ± 0.30%) compared to α⁺ (2.42 ± 0.29%), while HbF was mildly elevated in α⁰ (0.41 ± 0.35%) relative to α⁺ (0.13 ± 0.29%), which is in line with previous observations of increased HbF in α⁰ thalassemia due to erythropoietic stress32.
PLT indices, including PLT, PDW, MPV, and P-LCR, remained within normal ranges in both groups. However, α⁰ carriers displayed slightly higher mean values for PDW (13.96 ± 2.47%) and P-LCR (28.26 ± 6.75%) compared to α⁺ carriers.
For leukocyte indices, WBC counts were similar between α⁰ (6.95 ± 1.74 × 10³/µL) and α⁺ (7.31 ± 1.73 × 10³/µL) groups. Nuet% and Lym% showed comparable distributions, with no major clinical differences between groups, though α⁰ carriers exhibited slightly higher MXD% (9.13 ± 3.56%) than α⁺ (8.14 ± 2.97%).
To evaluate differences in hematological and electrophoresis parameters between individuals with α⁰-thalassemia and α⁺-thalassemia, we conducted a series of statistical comparisons. Following normality assessment using the D’Agostino–Pearson omnibus test, appropriate statistical tests were selected for each feature. For normally distributed variables with equal variances (assessed via Levene’s test), Student’s t-test was applied; for those with unequal variances, Welch’s t-test was used. Non-normally distributed features were compared using the Mann–Whitney U test. To account for the risk of false positives due to multiple hypothesis testing, p-values were adjusted using the Benjamini–Hochberg false discovery rate correction. Effect sizes (Cliff’s delta, δ) were calculated for each comparison and classified according to established benchmarks: negligible (< 0.15), small (0.15–0.33), medium (0.33–0.47), and large (≥ 0.47).
Out of the 19 evaluated hematologic features, 16 demonstrated statistically significant differences (adjusted p < 0.05) between α⁰- and α⁺-thalassemia carriers. Several parameters, including MCV, MCH, MCHC, and RDW-CVRL, demonstrated large effects (δ ≥ 0.47), indicating marked phenotypic separation and robust discriminative potential between the two genotypic groups. Additional parameters such as RBC, Hb, and HbF showed medium effects (0.33 ≤ δ < 0.47) with strong statistical significance, further supporting their role in differentiating α-thalassemia subtypes. Markers such as HBA, HBA₂, PDW, MPV, and P-LCR displayed small effects (0.15 ≤ δ < 0.33), suggesting modest but potentially relevant contributions to the phenotype.
In contrast, PLT, Lym%, and Nuet% showed no significant group differences (adjusted p > 0.05) and negligible effect sizes, indicating minimal diagnostic utility. Finally, WBC and MXD%, as well as Lym# were statistically significant but yielded negligible effect sizes (δ < 0.15), suggesting that while differences are detectable statistically, they may not translate into clinical relevance.
Taken together, these findings highlight a subset of hematologic indices, particularly red cell parameters and globin fractions as the most informative for distinguishing between α⁰- and α⁺-thalassemia carriers, with implications for both diagnostic strategies and the development of predictive models in clinical settings.
Data preparation
The dataset underwent a series of preprocessing steps to ensure data quality and suitability for machine learning analysis. The process began with dataset annotation by labeling each sample as either \(\:{\alpha\:}^{+}\) or \(\:{\alpha\:}^{0}\), in which six common large mutations associated with alpha-thalassemia were categorized into two classes: alpha-plus (\(\:{\alpha\:}^{+}\)) and alpha-zero (\(\:{\alpha\:}^{0}\)). Patients with heterozygous α3.7 or α4.2 mutations were labeled as alpha-plus, whereas those with − α20.5, −−Med, compound heterozygous α3.7/α4.2, and homozygous α4.2 were categorized as alpha-zero. The gender variable, as a categorical feature, was encoded using one-hot encoding to convert it into binary form for compatibility with machine learning models. Subsequently, irrelevant features such as Medical Record Number (MRN) were removed, as they lacked analytical significance, to enhance model efficiency and reduce potential noise. Next, data cleaning was performed to address common errors introduced during the manual transfer of data from paper-based medical records to a CSV file. Errors such as typographical mistakes, column or row shifting, format inconsistencies, and duplicate records, which occurred due to patients being referred to the center multiple times over the 22-year period, were detected and rectified to ensure data integrity. These preprocessing steps eliminated noise, irrelevant features, addressed data incompleteness, and optimized the dataset for subsequent machine learning applications.
Handling missing value
A critical aspect of data preprocessing involved handling missing values to prevent biases and inaccuracies in the analysis. For continuous features, including Hb, RDW-CVRL, HBA, Platelet, WBC, Lym%, MXD%, Nuet%, Lym#, MPV, and P-LCR, missing values were imputed using the K-nearest neighbors (KNN) algorithm. This method calculates Euclidean distances to identify the five nearest neighbors and replaces missing values with the average of these neighbors, thereby preserving data distribution patterns and ensuring consistency in data representation.
Outliers detection
In this study, the Local Outlier Factor (LOF)33 algorithm was employed to identify abnormal samples within the dataset. First, the dataset containing hematological parameters of α-thalassemia carriers was input into the model. The algorithm then computed the distances between each data point and its five nearest neighbors (n_neighbors = 5) using the Euclidean distance metric (p = 2). Following this, the local reachability density (LRD) for each sample was calculated based on these distances. The LOF score for each record was then derived by comparing its LRD with those of its neighboring points. Finally, data points with LOF scores exceeding a predefined threshold were marked as outliers and excluded from further modeling. A total of 18 outliers were detected and subsequently removed from the dataset, as their limited presence did not allow for meaningful analysis or reliable insights. This decision ensured that the model remained robust without being influenced by a small set of anomalous cases. This process is consistent with the general LOF methodology described by Zou et al. (2023) (Fig. 6)34 (Tables 3, 4).
Feature selection
To improve model performance, feature selection was conducted using the Extra Trees algorithm35, an ensemble method that builds multiple randomized decision trees. Unlike Random Forest, which uses bootstrap sampling and selects the best split based on impurity, Extra Trees uses the entire dataset without resampling and chooses split points randomly from a subset of features, reducing variance and improving generalization, especially for high-dimensional or noisy datasets36,37. Extra Trees constructs trees using the entire dataset without resampling and applies random splits for feature selection, increasing diversity and reducing variance37,38. A key advantage of Extra Trees is its built-in feature selection mechanism, where features are ranked based on their contribution to the reduction of impurity, a measure known as Gini Importance or Mean Decrease in Impurity. Features with higher scores are considered more relevant for the classification task, while those with lower importance can be discarded. This ranking process helps in dimensionality reduction, improving computational efficiency and preventing overfitting. The top-k most important features are often selected based on their ranking, ensuring that only the most relevant features are retained for model training38,39.
The process of tree construction in Extra Trees follows a systematic yet randomized approach. At each node, a random subset of features is selected, and multiple potential split points are randomly determined. The best split is then chosen from these randomly generated options, which helps prevent overfitting and enhances the model’s generalization ability. The splitting decision is typically based on an impurity criterion such as Gini Index or Entropy, ensuring an effective division of the dataset into homogeneous subgroups. This process continues recursively until a stopping criterion, such as a minimum number of samples per leaf node, is met40,41 Extra Trees Classifier provides several advantages over other tree-based methods. By incorporating randomness in split selection, it exhibits lower variance and is less prone to overfitting compared to models that select the best split deterministically. Additionally, its computational efficiency is higher than that of Random Forest since it does not perform an exhaustive search for the optimal split, making it faster when handling large datasets. Moreover, the ensemble nature of the model, where predictions are aggregated through majority voting for classification or averaging for regression, enhances overall stability and accuracy39,42. The blood parameters were evaluated through this approach to identify the most informative features for alpha-thalassemia classification. The performance of the Extra Trees algorithm is influenced by several hyperparameters that control tree growth, feature selection, and prediction stability. These hyperparameters, along with their respective values, are summarized in Table 5.
Modeling
General experimental framework
All four algorithms: Logistic Regression (LR), Support Vector Machines (SVM), KNN, and Gradient Boosting (GB) were implemented within a standardized scikit-learn pipeline incorporating z-score normalization via StandardScaler to ensure consistent feature scaling. Model development followed a nested stratified cross-validation (CV) strategy with an outer 10-fold CV for unbiased generalization estimation and an inner 5-fold CV for hyperparameter tuning via GridSearchCV. This procedure was repeated across five random seeds (42, 123, 456, 789, 2023) to minimize variance from data partitioning. Class imbalance between α⁺ and α⁰ carriers was addressed using class_weight=’balanced’ where applicable. Model selection within the inner loop prioritized the F1-score, balancing false positives and false negatives to reflect the clinical importance of both error types.
Following hyperparameter optimization,the best model from each outer fold underwent probability calibration using CalibratedClassifierCV (5-fold, training data only) to improve clinical interpretability of predicted risk scores. Sigmoid (Platt scaling) or isotonic regression was applied depending on model suitability. Performance was evaluated at a fixed 0.5 threshold using six metrics: accuracy, precision, recall, specificity, F1-score, and AUC-ROC (Receiver Operating Characteristic Area Under the Curve) with results reported as mean ± standard deviation across all folds and seeds. The final version of each model was retrained on the full dataset using the optimal hyperparameters and saved for reproducibility.
Model-specific details
-
LR:
-
Explored three regularization schemes: L1 (liblinear solver), L2 (lbfgs or saga solvers), and Elastic Net (saga solver) with l1_ratio ∈ {0.1, 0.5, 0.9}. The inverse regularization strength C spanned 10⁻⁴ to 10⁴ (logarithmic scale). The maximum number of iterations was set to 5000 to ensure convergence.
-
SVM:
-
Tested both linear and RBF kernels, with C ∈ {0.01, 0.1, 1, 10, 100}. For RBF, gamma ∈ {0.001, 0.01, 0.1, 1, ‘scale’, ‘auto’}. Probability estimation was enabled (probability = True) to support calibration and decision-threshold adjustment.
-
KNN:
-
Used distance-weighted voting (weights=’distance’) with n_neighbors ranging from 3 to 30 (odd integers), p ∈ {1, 2} for Manhattan/Euclidean metrics, and leaf_size ∈ {10, 20, 30, 50}. Search algorithms included {‘auto’, ‘ball_tree’, ‘kd_tree’, ‘brute’}.
-
GB:
Hyperparameter search included n_estimators ∈ {50, 100, 200}, learning_rate ∈ {0.01, 0.05, 0.1}, max_depth ∈ {3, 4, 5}, min_samples_split ∈ {2, 5, 10}, min_samples_leaf ∈ {1, 2, 4}, subsample ∈ {0.8, 0.9, 1.0}, max_features ∈ {‘sqrt’, ‘log2’, None}, and ccp_alpha ∈ {0, 0.001, 0.01}.
Stacking ensemble
The stacking ensemble was constructed to leverage the complementary strengths of four independently optimized base classifiers: GB, SVM, KNN, and LR, whose hyperparameters were fixed at the optimal values identified in prior single-model experiments.
Configurations were:
-
GB: learning_rate = 0.05, max_depth = 5, min_samples_leaf = 4, min_samples_split = 2, subsample = 0.8, n_estimators = 200, max_features = None, ccp_alpha = 0, random_state = 42.
-
SVM (RBF): C = 100, gamma = 0.001, kernel=’rbf’, probability = True, class_weight={0:w0,1:w1}, random_state = 42 within a StandardScaler() pipeline.
-
KNN: n_neighbors = 19, p = 1, weights=’distance’, leaf_size = 10, algorithm=’auto’, metric=’minkowski’ within a StandardScaler() pipeline.
-
LR (elastic net): C = 0.01, l1_ratio = 0.1, penalty=’elasticnet’, solver=’saga’, max_iter = 5000, class_weight={0:w0,1:w1}, random_state = 42 within a StandardScaler() pipeline.
Class imbalance between α⁺ and α⁰ carriers was handled in applicable models via class_weight derived from training-set class frequencies. The meta-learner was a LR model embedded in a StandardScaler() pipeline. Hyperparameters were optimized via grid search with 5-fold CV on out-of-fold meta-features using the search space:
-
penalty ∈ {‘l1’,‘l2’,‘elasticnet’}, C ∈ np.logspace(-4,4,9), solver ∈ {‘liblinear’,‘lbfgs’,‘saga’}, l1_ratio ∈ {0.1,0.5,0.9} (elastic net only), and class_weight ∈ {None,‘balanced’}; max_iter = 5000, random_state = 42.
To prevent information leakage into meta-training, meta-features were the base learners’ out-of-fold predicted probabilities computed with 5-fold CV on each outer-fold training partition. Model selection followed a nested stratified CV with an outer 10-fold (n_splits = 10, shuffle = True, random_state = 42). Within each outer training fold, base model out-of-fold predicted probabilities were first generated; the meta-learner was then tuned in an inner 5-fold CV with scoring=’f1’. The best meta-learner was integrated into a StackingClassifier(cv = 5, passthrough = False), and the ensemble’s probability outputs were calibrated using a sigmoid method (CalibratedClassifierCV(method=’sigmoid’, cv = 5)), fit only on the training partition of each outer fold.
For decision-making, metrics were reported at the default threshold (0.5) and at a per-fold optimized threshold chosen from the outer test fold precision–recall curve to maximize F1. This threshold selection does not affect model fitting, but uses test labels for selection; therefore, the 0.5-threshold results are treated as the primary unbiased estimates, while the optimized-threshold results are provided as complementary operating points. Performance on outer test folds includes accuracy, precision, recall, specificity, F1-score, and AUC-ROC (AUC is threshold-independent), summarized as mean ± standard deviation across outer folds. Finally, the calibrated stacking model was refit on the full dataset and serialized for reproducibility.
Evaluation
Performance metrics
The performance of all models in classifying α⁰ versus α⁺ thalassemia was assessed using multiple complementary metrics to ensure both statistical robustness and clinical reliability.
$$\:\text{Accuracy}=\frac{TP+TN}{TP+TN+FP+FN}\times\:100$$
(1)
While informative, accuracy alone can be misleading for imbalanced data, and therefore was interpreted alongside other metrics.
$$\:\text{Precision}=\frac{TP}{TP+FP}\times\:100$$
(2)
High precision reduces false positives (α⁺ misclassified as α⁰), minimizing unnecessary genetic testing and patient anxiety.
$$\:\text{Recall}=\frac{TP}{TP+FN}\times\:100$$
(3)
$$\:F1=2\times\:\frac{\text{Precision}\times\:\text{Recall}}{\text{Precision}+\text{Recall}}\times\:100$$
(4)
This is especially relevant given the trade-off between precision and recall, and the greater clinical harm of false negatives (α⁰ misclassified as α⁺) compared to false positives.
-
AUC (Area Under the ROC Curve) provided a threshold-independent measure of discriminative ability, summarizing the trade-off between true positive rate and false positive rate across all decision thresholds.
-
Here, the classification outcomes can be summarized in Table 6.

