Model training and optimization
To identify the optimal model regarding predictive accuracy and generalizability, fifteen ML classifiers were generated by integrating five distinct algorithms with three molecular descriptors (Table 1). Model performance was assessed via 5-fold stratified cross-validation applied to the training set. Among the assessed metrics, the Matthews Correlation Coefficient (MCC) was selected as the primary criterion for model comparison, with the resulting performance scores listed in Table 1.
As listed in Table 1, cross-validation unveiled mean MCC values spanning from 0.330 to 0.644. SVM and XGBoost algorithms consistently demonstrated outstanding performance across all three fingerprints, occupying the top six positions in the evaluation. XGBoost models were trained using ECFP4 and RDKit descriptors, yielding MCC values of 0.636 ± 0.103 and 0.628 ± 0.101, respectively. While no single fingerprint type dominated, the presence of MACCS, ECFP4, and RDKit representations among the top five performers indicated that each feature set provided distinct and valuable insights for predicting activity. Conversely, models utilizing LR demonstrated moderate performance levels, whereas KNN and Naïve Bayes classifiers consistently produced lower MCC values across all feature representations. Ultimately, the SVM model trained on MACCS keys demonstrated the highest predictive power, achieving a peak mean MCC of 0.644 ± 0.103. Consequently, this optimal classifier was applied to screen the NCI database.
Validation of the final model and virtual screening
Following hyperparameter optimization, the best-performing SVM model—based on MACCS fingerprints—was retrained on the full training dataset (Table 2). Its generalization ability was then evaluated using an independent hold-out test set. The classifier attained an MCC of 0.556 and an overall accuracy of 84.8%. As shown in Fig. 2a, the ROC-AUC reached 0.76, while the PRC-AUC was 0.69 (Fig. 2b), indicating a moderate ability to discriminate between active and inactive compounds. The combination of high precision and lower recall suggests that the model’s positive predictions are relatively reliable, although only a subset of the active chemical space is recovered. This trade-off is often advantageous in virtual screening workflows, where minimizing false positives among prioritized compounds may be more important than maximizing recall before applying computationally intensive methods.
For these reasons, the ML model was employed only as an initial ligand-prioritization tool rather than a standalone activity-confirmation method. The ML-predicted active compounds were subsequently filtered and validated through molecular docking, MD simulations, MM-GBSA binding energy calculations, and post-MD stability analyses. Accordingly, the identified NCI compounds should be regarded as promising candidates whose predictive reliability is supported by this multi-stage workflow, while experimental validation remains necessary to confirm their inhibitory activity.
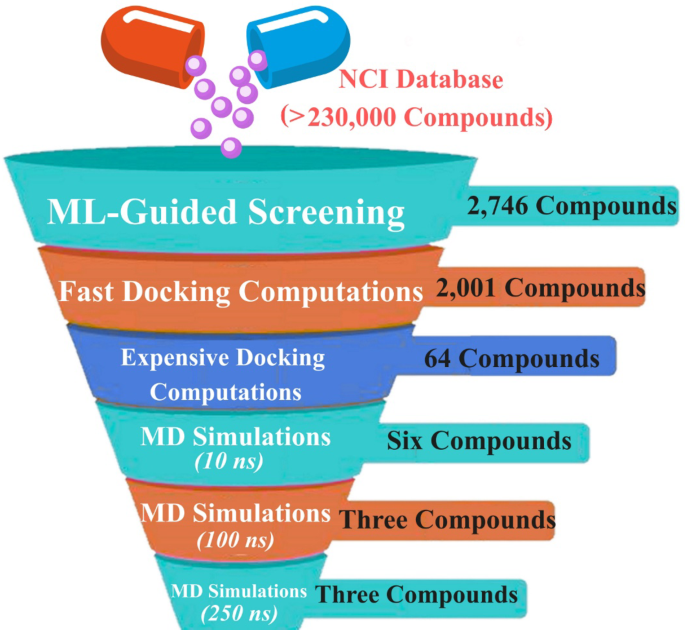
The virtual screening workflow was designed as a stepwise enrichment strategy to progressively prioritize the most promising NRP1-binding candidates from the large NCI chemical library. Starting from more than 230,000 NCI compounds, the final validated SVM model predicted 2,746 compounds as potentially active against NRP1. The predicted active compounds were subsequently investigated utilizing molecular docking to explore their potential binding modes and scores within the NRP1 active site.

Performance evaluation of the optimized SVM-MACCS model on the independent test set: (a) Receiver operating characteristic curve showing the discriminatory ability of the model between active and inactive NRP1 compounds and (b) Precision-recall curve illustrating the balance between precision and recall, which is particularly informative for evaluating imbalanced datasets.
Docking predictions
The reliability of the docking protocol for predicting inhibitor binding poses inside the NRP1 active site has been previously established in the literature51,65. Briefly, docking calculations were performed using AutoDock Vina1.1.2 software. To further validate the adopted docking setup, the co-crystallized HRG/Arg-1 ligand was redocked into the NRP1 active site. The superimposition of the predicted and native crystallographic binding poses is shown in Figure S1. The redocked pose of HRG/Arg-1 closely reproduced the native binding orientation, with an RMSD of 0.23 Å, well below the widely accepted threshold of 2.0 Å. This confirms the reliability of the docking protocol for reproducing the experimental binding pose within the NRP1 active site. Consequently, all ML models predicted that 2,746 potentially active NCI compounds were advanced for fast docking computations against NRP1 (Table S2). Among them, 2,001 NCI compounds unveiled docking scores lower than or equal to that of the reference ligand (HRG/Arg-1; calc. −5.7 kcal/mol). Consequently, those 2,001 NCI compounds were advanced for expensive docking computations. The outcomes of expensive docking calculations are listed in Table S3, where 1,983 NCI compounds unveiled docking scores less than or equal to HRG/Arg-1 (calc. −5.7 kcal/mol). To shortlist the NCI compounds, a threshold of −7.5 kcal/mol was used to identify potential NRP1 inhibitors, yielding 64 compounds. Table 3 displays the 2D chemical structures, intermolecular H-bonds, and the estimated docking scores of the three most potent NCI compounds against NRP1. Of note, three NCI compounds were selected based on MD simulations over 250 ns, as detailed in the subsequent sections.
Figure 3 illustrates the 3D and 2D visualizations of the anticipated docking modes of NCI704332, NCI717568, and NCI674566 within the NRP1 active site. Among them, NCI704332 revealed a superior docking score of − 7.8 kcal/mol and established five H-bonds with proximal residues of the NRP1 active site. More exactly, the NH and C = O moieties of 2-chloro-N-(4-fluorophenyl)-4-mercaptobenzamide participated in the formation of two H-bonds with the OH of TYR297 (3.14 Å) and the terminal amine of LYS351 (2.02 Å), respectively. Furthermore, the S = O group of (R)-4-chloro-2-mercapto-N-(6-methyl-1,4,5,6-tetrahydro-1,2,4-triazin-3-yl)benzenesulfonamide interacted via H-bonding with the hydroxyl hydrogen of TYR297 (2.85 Å) and the amide nitrogen of TRP301 (2.37 Å). Additionally, the NH of the same moiety in the latter formed an H-bond with the C = O of GLU348 (2.11 Å), contributing to the overall stabilization of the NCI704332-NRP1 complex.
NCI717568 was found to bind favorably to the NRP1 active site, with a docking score of −7.6 kcal/mol. The nitrogen atom of the 1-(4-chlorobenzyl)-1 H-benzo[d]imidazole scaffold participated in two H-bonding interactions with the amide NH of TRP301 (2.21 Å) and the hydroxyl of SER346 (2.99 Å), enhancing the stability of the NCI717568-NRP1 complex (Fig. 3).
Finally, NCI717566 also showed a favorable docking score of −7.6 kcal/mol and formed three H-bonds with key residues within the NRP1 active site (Fig. 3). Minutely, the C = O of 2,3-diphenylquinazolin-4(3H)-one engaged in two H-bonds with the NH of TRP301 (1.17 Å) and the NH2 of ASN313 (1.99 Å). In addition, the C = O group of (S)-2-(3-chlorophenyl)-3-phenylthiazolidin-4-one interacted via an H-bond with the OH group of TYR353 (1.94 Å).

Predicted binding modes and protein-ligand interactions of the top-ranked NCI compounds within the NRP1 active site. The figure shows the 3D binding orientations and 2D interaction maps of (a) NCI704332, (b) NCI717568, and (c) NCI674566, highlighting the key H-bonding and noncovalent interactions with NRP1 active-site residues.
MD simulations
MD simulations were applied to explore the interaction dynamics and stability of the identified NCI compounds with NRP1. Therefore, 64 top-ranking NCI compounds with docking scores below −7.5 kcal/mol were initially simulated for 10 ns, and their ΔGbinding were evaluated (Table S4). As listed in Table S4, six out of 64 NCI compounds disclosed superior binding affinities than the HRG/Arg-1 (ΔGbinding = −29.8 kcal/mol). These six NCI compounds were further subjected to longer MD throughout a simulation time of 100 ns, accompanied by binding energy evaluations (Table S5). As shown in Table S5, three compounds, namely NCI704332, NCI717568, and NCI674566, disclosed ΔGbinding values less than HRG/Arg-1 (ΔGbinding = −31.2 kcal/mol).To achieve more trustworthy results, these three NCI compounds were introduced to 250 ns MD simulations, along with their binding energy evaluations (Fig. 4). Upon the MM-GBSA//250 ns MD simulations, NCI704332, NCI717568, and NCI674566 demonstrated promising ΔGbinding compared to HRG/Arg-1 throughout 250 ns MD simulations, with values of −36.7, −32.5, −31.9, and −30.2 kcal/mol, respectively (Fig. 4). Collectively, these results shed new light on NCI704332, NCI717568, and NCI674566 as potential inhibitors with high binding stability toward NRP1.
Despite these promising computational findings, the present study is limited by its exclusive reliance on in-silico methods, and the identified NCI compounds should therefore be regarded as potential candidates that require experimental validation. Future work should first evaluate the direct binding and inhibitory activity of NCI704332, NCI717568, and NCI674566 against NRP1 through in-vitro biochemical assays, such as ligand-binding, competition, or fluorescence-based assays. Compounds confirmed to bind should then be assessed in cell-based experiments using NRP1-overexpressing cancer cell lines to determine their effects on cancer-related phenotypes, including viability, migration, invasion, and VEGF/NRP1-mediated signaling. The most promising candidates may subsequently be advanced to in-vivo tumor models to investigate their pharmacokinetic profiles, anticancer efficacy, and preliminary safety. Such validation will be essential to confirm the biological relevance of the computational predictions and to guide subsequent lead optimization.

MM-GBSA binding energy evaluation of NCI704332, NCI717568, NCI674566, and the reference ligand HRG/Arg-1 in complex with NRP1. Binding energies were calculated over 10, 100, and 250 ns MD simulations to assess the persistence of ligand binding affinity over increasing simulation times.
To gain a deeper understanding of the driving forces responsible for target-ligand binding, the estimated binding energies of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 complexed with NRP1 were decomposed into their individual components (Fig. 5). As depicted in Fig. 5, ΔEvdW and ΔEele interactions were the dominant participants in complex stabilization over 250 ns MD simulations. Specifically, NCI704332, NCI717568, NCI674566, and HRG/Arg-1 bound to NRP1 demonstrated ΔEvdW values of −37.0, −44.1, −45.4, and −16.5 kcal/mol. In addition, the ΔEele values were −51.6, −18.1, −19.1, and −164.3 kcal/mol for NCI704332-, NCI717568-, NCI674566-, and HRG/Arg-1-NRP1 complexes, respectively. These data highlighted the predominance of van der Waals forces in stabilizing NCI704332, NCI717568, and NCI674566, whereas electrostatic interactions disclosed a more significant influence on the binding of HRG/Arg-1 with NRP1.

Decomposition of MM-GBSA binding-energy components for NCI704332, NCI717568, NCI674566, and HRG/Arg-1 bound to NRP1 over 250 ns MD simulations. The plotted terms include electrostatic, van der Waals, polar solvation, and nonpolar solvation contributions, illustrating the major energetic factors stabilizing each protein-ligand complex.
Furthermore, per-residue decomposition analysis was executed to determine which residues primarily drive the interactions of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 with NRP1 (Fig. 6). Only residues with ΔGbinding < −0.5 kcal/mol were considered. TRP301 displayed a consistently strong contribution with ΔGbinding values of −2.8, −4.1, −3.1, and −1.4 kcal/mol for NCI704332-, NCI717568-, NCI674566-, and HRG/Arg-1-NRP1 complexes, respectively (Fig. 6). These observations underscored the importance of TRP301 in stabilizing inhibitor interactions within the NRP1 active site.

Per-residue MM-GBSA decomposition analysis of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 in complex with NRP1 over 250 ns MD simulations. Only residues contributing favorably to ligand binding are shown, highlighting the key NRP1 residues involved in stabilizing the identified compounds within the active site.
Post-MD analyses
Binding energy per trajectory
To evaluate the energetic stability of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 bound to NRP1, a binding energy per trajectory analysis was executed over 250 ns MD simulations (Fig. 7a). As disclosed in Fig. 7a, the NCI704332, NCI717568, and NCI674566 in complex with NRP1 manifested relatively steadiness with ΔGbinding values of −36.7, −32.5, and −31.9 kcal/mol, respectively, compared to HRG/Arg-1 (calc. −30.2 kcal/mol). These results indicated that the ligand remained associated with the NRP1 binding site over the course of the simulation, supporting the stability of the complex.
Center-of-mass (CoM) distance
To further understand the structural stability of inhibitor-target interactions, the CoM distances between NCI704332, NCI717568, NCI674566, and HRG/Arg-1 and TRP301 within the NRP1 active site were gauged throughout 250 ns MD simulations (Fig. 7b). The outcomes unveiled that NCI704332, NCI717568, and NCI674566 maintained close and stable interactions with TRP301, with average CoM distances of 5.7, 7.3, and 6.4 Å, respectively. The HRG/Arg-1 complexed with NRP1 revealed a mean CoM distance of 6.2 Å, implying a comparable degree of binding proximity. These results supported the persistence of the ligand within the NRP1 active site throughout the simulation period.
Root-mean-square deviation (RMSD)
To inspect the conformational changes and structural stability of the investigated complexes, RMSD analysis was performed for NCI704332, NCI717568, NCI674566, and HRG/Arg-1 bound to NRP1. The backbone RMSD values of each complex were calculated relative to their initial conformations over 250 ns MD simulations (Fig. 7c). The average RMSD values for the NCI704332-, NCI717568-, NCI674566-, and HRG/Arg-1-NRP1 complexes were approximately 0.2 nm. These findings proposed that all inspected compounds exhibited tight binding with NRP1 and induced minimal structural perturbations, thus preserving the overall conformational stability of the receptor.

Post-MD stability analyses of NCI704332 (purple), NCI717568 (mint green), NCI674566 (gray), and HRG/Arg-1 (blue) bound to NRP1 over 250 ns MD simulations: (a) binding energy per trajectory analysis showing the stability of ligand binding, (b) CoM distances between each ligand and TRP301, and (c) backbone RMSD profiles of the protein-ligand complexes, reflecting the overall structural stability of NRP1 during the simulations.
Root-mean-square fluctuation (RMSF)
To characterize the local mobility and flexibility of NRP1 residues, RMSF analysis was executed for the apo-NRP1 and its structure complexed with NCI704332, NCI717568, NCI674566, and HRG/Arg-1. The RMSF analysis revealed comparable fluctuations of approximately 0.1 nm for apo-NRP1, NCI704332-, NCI717568-, and HRG/Arg-1-NRP1 complexes, whereas the NCI674566-NRP1 complex showed slightly higher residue flexibility, averaging 0.2 nm (Fig. 8a). Overall, the low RMSF values indicated that ligand binding did not markedly perturb NRP1 flexibility during the 250 ns MD simulations.

Structural flexibility and compactness analyses of apo-NRP1 (black) and NRP1 complexed with NCI704332 (purple), NCI717568 (mint green), NCI674566 (gray), and HRG/Arg-1 (blue) over 250 ns MD simulations: (a) RMSF profiles showing residue-level flexibility of NRP1 in the apo and ligand-bound states, and (b) Rg profiles assessing the compactness and folding stability of NRP1 throughout the simulations.
Radius of gyration (Rg)
The Rg analysis was conducted for NCI704332, NCI717568, and NCI674566, and the HRG/Arg-1 complexed with NRP1 throughout 250 ns MD simulations to inspect how residue flexibility affects protein compactness (Fig. 8b). From Fig. 8b, the measured average Rg values for apo-NRP1 and compound-NRP1 complexes were almost identical, with a value of 1.5 nm. These results suggested that ligand binding did not alter the overall compactness of NRP1.
Drug-likeness properties
Ro5 was utilized to estimate the oral bioavailability and drug-likeness of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 utilizing the SwissADME web tool (Fig. 9). The anticipated Mlog P values for NCI704332, NCI717568, and NCI674566 were found to be 2.71, 5.89, and 6.36, while their MWs were 457.93, 567.89, and 586.10 g/mol, respectively. HRG/Arg-1 exhibited markedly lower values, with a Mlog P of −0.94 and a MW of 191.25 g/mol. Consistent with Ro5 parameters, NCI704332 achieved the lipophilicity and MW requirements, while NCI717568 and NCI674566 slightly exceeded these limits. The number of HBA/HBD for NCI704332, NCI717568, NCI674566, and HRG/Arg-1 were 5/4, 2/0, 3/0, and 3/6, respectively, fell within acceptable ranges. Altogether, these outcomes ensured that all identified NCI compounds displayed reasonable drug-like characteristics, with NCI704332 emerging as the most Ro5-compliant candidate for potential inhibition of NRP1.
It is worth noting that NCI717568 and NCI674566 exceeded Lipinski’s recommended limits for both MW and lipophilicity, with MW values of 567.89 and 586.10 g/mol and Mlog P values of 5.89 and 6.36, respectively. Each, therefore, incurred two violations of Lipinski’s Ro5. These deviations may adversely affect aqueous solubility, passive membrane permeability, and oral bioavailability. Although these two compounds exhibited favorable docking scores, binding energies, and dynamic stability within the NRP1 active site, further structural optimization may be needed to improve their physicochemical and pharmacokinetic profiles. In contrast, NCI704332 showed full compliance with Lipinski’s Ro5, suggesting that it may represent the most drug-like candidate among the identified NCI compounds.

Predicted drug-likeness properties of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 according to Lipinski’s Ro5. The evaluated descriptors include (a) Mlog P, (b) molecular weight, (c) number of HBA, and (d) number of HBD, which were used to assess the oral drug-likeness potential of the identified compounds and HGR/Arg-1.
ADME features
To estimate their pharmacokinetic suitability, the ADME characteristics of NCI704332, NCI717568, and NCI674566 were anticipated and compared to those of HRG/Arg-1 (Table 4). Under the absorption category, Caco-2 permeability and HIA values revealed enhanced absorption potential for all NCI compounds relative to HRG/Arg-1. The evaluated Caco-2 values were −0.135, 1.009, 1.064, and −0.351, while HIA values were 69.46%, 89.95%, 100%, and 24.6%, respectively. For distribution, the VDss was greatest for NCI704332 (calc. 1.109) and lowest for HRG/Arg-1 (calc. −0.438), indicating better systemic exposure for the NCI compounds. Concerning metabolism, none of the investigated compounds were recognized as CYP1A2 inhibitors, suggesting limited interference with phase I metabolic enzymes. In terms of excretion, total clearance was moderate for all identified NCI compounds, with values ranging from 0.103 to 0.139; however, HRG/Arg-1 demonstrated a slight higher total clearance with a value of 0.589. Altogether, the predicted ADME features highlighted the promising pharmacokinetic potential of NCI704332, NCI717568, and NCI674566, particularly when compared with HRG/Arg-1, reinforcing their suitability as putative NRP1-targeted anticancer agents.
Quantum mechanical (QM) calculations
QM computations were executed on the final snapshots of NCI704332, NCI717568, NCI674566, and HRG/Arg-1, extracted from 250 ns MD simulations. The MEP maps were generated and plotted for the optimized structures of NCI704332, NCI717568, NCI674566, and HRG/Arg-1 to visualize their nucleophilic and electrophilic regions (Fig. 10). As depicted in Fig. 10, the electronegative regions shown in red were observed around the N and O atoms of the inspected compounds, corresponding to potential H-bond acceptor sites. These regions are consistent with the docking results, in which the carbonyl, sulfonyl, and heteroatom-containing groups of the identified compounds participated in H-bond interactions with key NRP1 residues, including TRP301, TYR297, GLU348, LYS351, SER346, ASN313, and TYR353. In contrast, the electropositive regions shown in blue, mainly located around H atoms attached to heteroatoms or aromatic frameworks, represent potential hydrogen-bond donor or electrostatic interaction sites. Therefore, the MEP distributions supported the docking-predicted interaction patterns and provided an electronic rationale for the favorable binding orientations of NCI704332, NCI717568, and NCI674566 within the NRP1 active site.

MEP maps for the optimized structures of (a) NCI704332, (b) NCI717568, (c) NCI674566, and (d) HRG/Arg-1. MEP colour ranges from −0.01 au (red) to + 0.01 au (blue).
Furthermore, the frontier molecular orbital (FMO) parameters, including EHOMO and ELUMO, were estimated for optimized structures of NCI704332, NCI717568, NCI674566, and HRG/Arg-1, followed by EFL and Egap estimation (Table 5). The spatial distributions of HOMO and LUMO orbitals for the NCI704332, NCI717568, NCI674566, and HRG/Arg-1 are shown in Fig. 11. According to Fig. 11, HOMO densities were mainly located around the electron-rich zones (i.e., O and N atoms) of the inspected compounds, whereas the LUMO densities were localized around the electron-deficient regions (i.e., C and H atoms). The computed Egap values for NCI704332, NCI717568, NCI674566, and HRG/Arg-1 were 5.8, 5.5, 6.5, and 7.6 eV, respectively (Table 5). The calculated Egap values demonstrated that HRG/Arg-1 had the largest value, suggesting its superior stability and minimal chemical reactivity compared to the other investigated NCI compounds. Moreover, the smaller Egap of the identified NCI compounds relative to HRG/Arg-1 suggests higher chemical reactivity, which may promote charge-transfer and polarization contributions to noncovalent binding within the NRP1 pocket, in agreement with the docking and MM-GBSA findings.

The spatial distribution of HOMO and LUMO of (a) NCI704332, (b) NCI717568, (c) NCI674566, and (d) HRG/Arg-1. The HOMO and LUMO distributions illustrate the electron-donating and electron-accepting regions of each compound, respectively, providing insight into their electronic reactivity and potential interaction sites.
To gain more reliable insights into electronic features, global descriptors were estimated for NCI704332, NCI717568, NCI674566, and HRG/Arg-1 (Table 5). As listed in Table 5, NCI704332, NCI717568, NCI674566, and HRG/Arg-1 revealed IP values spanning from 6.8 to 8.3 eV. Moreover, the EA values were 1.37, 1.38, 0.97, and 0.2 eV for NCI704332, NCI717568, NCI674566, and HRG/Arg-1, respectively. Furthermore, η and S values for NCI704332, NCI717568, NCI674566, and HRG/Arg-1 were in the range of 2.7 to 3.8 eV and 0.27 to 0.37 eV–1, respectively. Additionally, the ω values were found to range from 2.62 to 3.14 eV for the identified NCI and HRG/Arg-1. Together, these descriptors indicated that the identified compounds possessed electronic features favorable for noncovalent interactions with NRP1.


