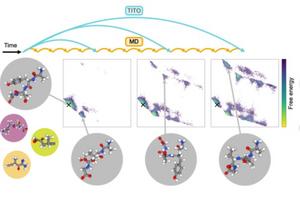
image:
TITO AI models learn to fast-forward through time at a faster rate than traditional numerical simulations, allowing researchers to characterize the physical properties of molecules more quickly. This model paves the way for rapid testing of new drugs in the future.
view more
Credit: Chalmers University of Technology |Juan Viguera Diez and Simon Olson
New AI models are very good at predicting how molecules will evolve over time, so they could potentially speed up the expensive and time-consuming testing process for new drugs in the future. In the long term, this technology could accelerate the development of medicines and new treatments by enabling faster and more accurate identification of promising drug candidates. The findings are published in a new Swedish study published in Science Advances.
Developing a new drug often takes more than 10 years from the initial idea to the time the finished drug reaches patients. Because a large number of tests must be performed to identify the most promising candidates, most of the associated costs and time are concentrated in the early stages. Multiple studies are often required to screen thousands of molecules, but only a small number of them move on to the next step.
Traditionally, the motion of molecules has been simulated using a technique known as molecular dynamics, in which researchers calculate the forces between all the atoms in steps, moving them a little at a time. For the calculation to be stable, each step must be very short, about 1 femtosecond (10⁻¹5 seconds). Processes critical to drug development occur over much longer timescales, requiring billions of steps and requiring much more computational power for simulation.
Big changes brought about by AI
The use of AI has enabled researchers to detect changes in molecules without having to perform numerical calculations. Machine learning can speed up each step of the calculation and directly generate plausible molecular structures without using generative models to simulate molecular motion.
A group of researchers from Chalmers University of Technology and the University of Gothenburg in Sweden has taken a new step forward by developing a new AI model that could make drug development trials even more efficient in the long run. The new model is more than 10,000 times faster than traditional simulations.
“Our AI What makes the model different is that it learns the underlying dynamics over longer timescales. It not only provides insight into the shapes that molecules take, but also how quickly and through which pathways these molecular transitions occur. As far as we know, this is the first time this has been done in a way that works for many different molecules,” said study leader and associate professor Simon Olsson from the Department of Computer Science and Engineering at Chalmers University of Technology and the University of Gothenburg.
Thousands of molecules have been tested
More than 12,500 organic molecules were investigated in this study, including organic molecules containing carbon, nitrogen, hydrogen, and oxygen atoms. More than 1,000 short peptides (molecules made of short chains of amino acids that make up proteins) were also studied. Because the AI model learned how molecules normally behave, it was able to fast-forward through the simulation. The results are still consistent with the laws of physics.
“We train the model using simulated examples of how atoms in molecules move over time. Based on these sequences, the model learns the underlying rules governing the movement of molecules and can predict how new molecules will behave,” said Simon Olsson.
The researchers compared the model’s results and conclusions with previous research on molecular evolution.
“We validated the results using extensive post-processing simulations to corroborate them using standard numerical algorithms, and they are consistent with each other,” says Simon Olson.
change is predictable
Although the AI model is not based on real images, the researchers describe the result as a way of jumping between scenes in a “molecular movie” rather than looking at every frame in sequence.
AI models form the basis of computational predictions that researchers make in the laboratory.
“So we measure very specific things: properties of the molecule, how ‘happy’ the molecule is in a particular solution, or, for example, whether the molecule wants to cross the membrane and enter the cell. But this is still in the future. ” says Simon Olson.
One major strength is that the model does not memorize individual systems, but instead learns the general rules that govern the motion of molecules, allowing the model to be applied to molecules it has never encountered before during training.
“There are certain patterns that models help identify. AI models are based on large numbers of examples and only observe what happens over a period of tens of nanoseconds. They are nevertheless able to predict the properties and changes in molecules that occur over a period of time a thousand times longer. Therefore, with the help of artificial intelligence, we can unravel what will happen in the ‘molecular future’.” “We can predict how molecules will change without ever seeing the process unfold,” says Simon Olson.
interesting for the pharmaceutical industry
“To be able to predict the physical phenomena exhibited by molecules, we need to understand the fundamental physics of how the system behaves. We believe we are among the first to demonstrate this in a general sense and show that it is possible,” said Juan Viguera Diez, an industrial PhD student at AstraZeneca in the Department of Computer Science and Engineering at Chalmers and Gothenburg University, and lead author of the paper.
Researchers see significant interest from industry in simulations that more accurately reflect reality and enable faster development of new drugs. The new AI model can speed up molecular simulations that require testing large numbers of potential molecules, so the research team hopes this will be an important step toward more efficient drug development.
“In the long term, AI models like ours could help identify promising drug candidates faster and improve accuracy at early stages. This research study shows what is possible now. This could pave the way for the development of more general technologies, and ultimately accelerate the development of new drugs and new treatments, as well as improve our understanding of disease in a broader sense,” says Juan Viguera Díez.
AI model details
The TITO (Transferable Implicit Transfer Operators) AI model is a deep generative modeling framework that learns the statistical rules governing the motion of molecules directly from simulation data. This makes it possible to predict how the atomic configuration (the way atoms are spatially arranged and related to each other within a molecule) changes over time scales much more quickly than traditional numerical simulations.
This method is currently being tested in a simplified solvent model and in small molecule systems at specific temperatures. Further development is currently underway for more complex and realistic systems.
Research details:
The article Transferable Generative models bridging molecular dynamics from femtosecond to nanosecond time steps was published in Science Advances. The authors are Juan Viguera Diez, Mathias Schreiner, and Simon Olsson, all from Chalmers University of Technology and the University of Gothenburg.
For more information, please contact us below.
Simon Olsson, Associate Professor, Department of Computer Science and Engineering at Chalmers University of Technology and University of Gothenburg, Sweden, simonols@chalmers.se
Simon Olsson speaks English, Danish, and German. Chalmers has an on-site podcast studio and filming equipment and can accommodate requests for TV, radio and podcast interviews.
Illustration caption: TITO AI models learn to fast-forward through time at a faster rate than traditional numerical simulations, allowing researchers to characterize the physical properties of molecules more quickly. This model paves the way for rapid testing of new drugs in the future.
Illustration credit: Chalmers University of Technology | Juan Viguera Diez and Simon Olson
Banner image caption: Researchers’ new AI models could ultimately pave the way for faster testing in drug development. The new model is up to approximately 10,000 times faster than traditional simulations.
Banner image credit: CC by p_a_h
Research method
Computational simulation/modeling
Research theme
not applicable
Article title
Transferable generative models bridging femtosecond to nanosecond timestep molecular dynamics
Article publication date
April 8, 2026
Conflict of interest statement
The authors declare that they have no competing interests.


