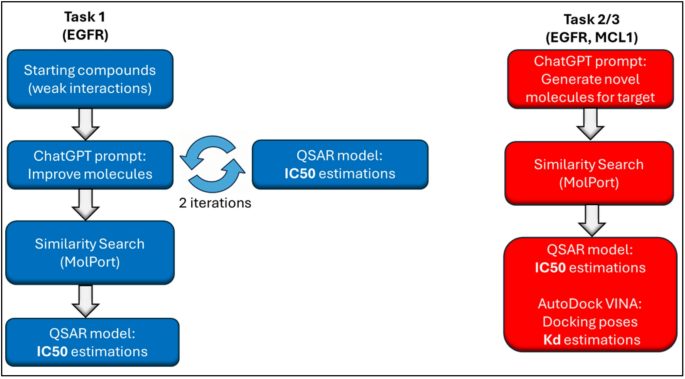
Task 1: Drug design of EGFR using an iterative procedure
In Task 1, three initial batches of compounds were provided to ChatGPT. Each batch contained five structurally diverse molecules with moderate activity values (pChEMBL: 5.01–5.47, corresponding to IC).50 values approximately 3–10 µM). These batches contained scaffolds and functional groups common to kinase inhibitors, such as halogenated aromatics, heterocycles, and polar groups such as nitriles and hydroxyls. The molecular structures and SMILES code details of these compounds are shown in Table S1 and Table S2. This diversity provided a wider chemical space for ChatGPT to explore.
Three independent experiments were conducted to evaluate the ability of ChatGPT to iteratively optimize the molecular design of EGFR using QSAR-based predictions. The QSAR model used for evaluation was constructed using available small molecule ICs50 Measurements for EGFR in ChEMBL (see Methods). Each experiment consists of two replicates, and the top predicted molecules after each batch and the last replicate (iteration 2) of the experiment are summarized in Table 1, Table S3, and Table S4.
The molecular structures of the top predicted compounds (9 compounds in total) from each batch and experiment are shown in Table S3, and the corresponding SMILES strings are shown in Table S4. These molecules have significantly improved predicted binding affinities compared to the starting compounds.
Remarkably, the generated molecules exhibited structural diversity across the backbone and functional groups, such as halogenated aromatics, heterocycles, and polar substituents, contributing to improved binding affinity predictions.
ChatGPT appears to extract important scaffolds from poorly performing initial molecules. Several of the generated molecules share features with known EGFR inhibitors, such as 4-quinazolineamine and benzimidazole derivatives, and halogenated aromatic rings, all of which are consistent with the pharmacophore of known EGFR inhibitors, indicating that the generation process of ChatGPT reflects an established structure-activity relationship.22, 23, 24, 25.
Some molecules feature unconventional elements for EGFR inhibitors, such as a thiourea-containing heterocycle (mol 1.6), a thiophene with multiple substituents including bromine and fluoro groups (mol 1.7), and a sulfonic group (mol 1.9). These atypical structures may increase chemical diversity and enable the search for new binding modes and resistant EGFR mutants.
This combination of structurally well-known and unusual scaffolds highlights ChatGPT’s ability to leverage known functionality while proposing new approaches to molecular design.
Similarity search and verification of predicted molecules
We evaluated the utility of GPT-generated molecules by performing a similarity search of the MolPort catalog (see Methods). This search identified commercially available compounds that were most similar to the highest affinity prediction from the final iteration based on the QSAR guide. The top ranked analogs were evaluated with a QSAR model and confirmed the predicted strong affinity. The three molecules identified belonged to the class of 4-quinazolineamine derivatives, which are well-established scaffolds for EGFR inhibition.
All three compounds are experimentally confirmed EGFR binders and support GPT design molecules as valid starting points for optimization. The structural similarity between the identified compounds and GPT predictions indicates that the generic model can converge to known pharmacologically relevant scaffolds without target-specific training. The structures of these identified 4-quinazoline amine derivatives and their respective predicted binding affinities are shown in Table 2.
Non-quinazoline derivatives identified by similarity search
In addition to quinazoline amine derivatives, MolPort searches revealed structurally diverse non-quinazoline compounds that resemble GPT design molecules. Four compounds (MOL 1.16–1.19, Table 3) represent opportunities for scaffold hopping in kinase inhibitor development.
Molecules 1.16–1.18 have no previous reports of EGFR inhibition, making them interesting candidates for experimental validation. Molecule 1.19 is associated with a protein kinase C (PKC) inhibitor patent26. PKC shares an ATP-binding domain with EGFR, potentially resulting in cross-reactivity, but kinase-specific differences highlight the need for careful evaluation.
Molecules 1.16–1.18 are characterized by unique structural motifs. Molecule 1.17 is derived from the malaria drug discovery library (CHEMBL5016744). Its structural motif may provide interesting opportunities for kinase inhibition.
In conclusion, ChatGPT produced molecules with improved affinity for EGFR. Similarity searches demonstrated that these outputs can guide the selection of viable candidates for experimental follow-up and address synthesizability limitations. This system proposed both known scaffolds such as 4-quinazoline amine derivatives and novel hinge-bound structures suitable for ATP-competitive kinase inhibition. Compounds such as 1.15-1.17 highlight testing opportunities, while 1.19 may exhibit cross-kinase activity. Overall, these results demonstrate the ability of ChatGPT to expand the chemical domain, explore both familiar and novel scaffolds, and support early-stage drug discovery.
Task 2: Design novel EGFR inhibitors and identify strong hits by similarity search
For Task 2, ChatGPT, as a drug development expert, was inspired to design five novel molecules with predicted strong affinity for EGFR. The aim was to propose a structurally different starting point that emphasizes novelty, druggability, and synthetic feasibility (Appendix A).
For each of the three experiments, the top molecules predicted by QSAR had estimated ICs.50 Values are 94 nM, 116 nM, and 338 nM (Table 4). Those SMILES were used in MolPort to identify the 10 most similar commercially available analogs per molecule and also evaluate their predicted binding affinities (Appendix B). Top analogs showed predicted IC50 Values of 55 nM, 56 nM, and 165 nM were obtained in each of the three experiments (Table 4), demonstrating that similarity searches can discover structurally diverse compounds with comparable or improved predicted binding affinities.
Next, we used AutoDock Vina to dock the top analogs and assess EGFR interactions. Docking estimated binding affinity (Kd) 1.35 μM, 10 nM, and 77 nM for the main compound in each experiment, indicating strong binding affinity (Table 4).
Of note, the best performing molecule, Mol 2.5, corresponds to omilinochol, an FDA-approved compound with no reported EGFR activity.27achieve the estimate Kd 10nM. The resulting docking pose suggests that its extended conformation reaches deep into the EGFR binding pocket (Figure 2). The benzimidazole group at one end of the molecule is located near hydrophobic residues Phe856 and Met766, which are important contacts for known EGFR inhibitors.28On the other hand, the benzimidazole group at the opposite end could bend onto the pocket surface and stabilize the ligand through hydrophobic interactions. Pyridine could form a potential hydrogen bond with Thr854, a key residue involved in ligand binding. The secondary amines are close to Asp855 (3.69 Å) and Thr845 (3.28 Å), suggesting additional hydrogen bonds.29. A central ketone-piperazine-ketone motif bridges the hydrophobic and polar regions, providing amphipathic flexibility. Piperazine has been used in several EGFR inhibitors to improve solubility and selectivity.30,31. It is important to note that our docking experiments do not take into account the effects of explicit water molecules, protein flexibility, and solvent, which may affect binding predictions. Overall, the extended mirror-like configuration of the benzimidazole in Mol 2.5 seems to maximize interactions within the pocket, suggesting good compatibility and a promising candidate for further optimization.

Docking simulation of molecule 2.5 in the EGFR binding pocket (3BEL). (a) Surface representation of the EGFR structure with molecule 2.5 (omilanochol) entering the binding pocket. (B) Close-up view of hydrophobic residues (ALA743, VAL726, LEU718) just after passing through the entrance. Also highlighted are the hydrogen bond with Thr854 (blue dotted line) and the estimated distance of a potential hydrogen bond with Asp855 of 3.694 Å (yellow dotted line). (C) Overall view of the molecule extending deep into the EGFR binding pocket. We demonstrate its unique 3D structure and interactions with important residues, including hydrophobic and hydrogen bond interactions.
Overall, the molecules generated by ChatGPT in task 2, combined with similarity searches, generated structurally diverse and easily accessible candidates with competitive predicted affinities for EGFR inhibition. See Appendix A and B for an overview of all generated compounds and their predicted activities, as well as the top 10 similar molecules and their similarity scores retrieved from MolPort.
Task 3: Design of novel non-covalent inhibitors of MCL1
In Task 3, we used ChatGPT to design a non-covalent inhibitor of MCL1, a BCL2 family protein associated with poor tumor outcomes.32. Five experiments were performed, each with five molecules proposed. From these experiments, six compounds have an estimated -9 kcal/mol (Kd=250 nM) or higher (Table S6).
A similarity search against MolPort yielded the top five most similar compounds for each of the six molecules (Appendix B). The top-ranked analogs have estimated binding affinities between -10.5 and -9.0 kcal/mol (Kd~10-250 nM) (Table 5).
A QSAR model was built using approximately 1000 compounds with known KI Values from ChEMBL. Mol 3.6, the molecule with the highest estimated docking affinity, has a concentration of approximately 1800 nM (KI). The highest scoring conformation of this docked ligand revealed that the quinazoline core is located near the entrance of the binding pocket, with two hydrophobic benzene rings extending into the highly hydrophobic region of the cavity (Figure 3). Another hydrophobic benzene ring near the entrance is thought to interact with the hydrophobic residues on the surface, improving the overall compatibility.
Attempts were made to introduce a functional group that could form a hydrogen bond with Arg263, a key residue in the MCL1 binding site, while maintaining a good fit within the hydrophobic pocket. ChatGPT was used to generate 20 linkers that connect the core structure to functional groups that can facilitate this interaction (Appendix A). The three linkers show significant improvement in QSAR prediction, with predicted KI The value is less than 100 nM. One linker formed a hydrogen bond with Arg263 and stabilized the complex (Figure 3C). QSAR predicted KI improved from approximately 1 μM to 46 nM, but the docking affinity remained good at −9.5 kcal/mol (Kd ~108 nM), providing a reasonable basis for further exploration.

Docking simulation of molecule 3.6 in the MCL1 binding pocket. (a) Intercalation of molecule 3.6 into the helix of the MCL1 structure (6FS1). (B) This molecule is tightly positioned in a hydrophobic pocket in close proximity to the key amino acids MET231 and VAL253. (C) Overlap of molecule 3.1 and its linker-modified form (-OCCCOC). The hydrogen bond to ARG263 is highlighted (dashed blue line). (D) Surface representation of MCL1 with both the original molecule (blue) and linker-modified molecule (green) superimposed on the binding pocket.


