image:
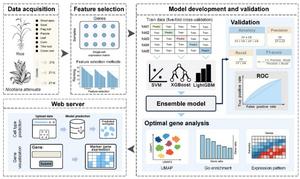
Figure 1 – Workflow of the proposed PhytoCell framework
view more
Credit: Qiang He
The rapid development of single-cell RNA-sequencing (scRNA-seq) has enabled researchers to examine gene activity in individual plant cells with unprecedented resolution, opening new opportunities to study cell differentiation, tissue development, and stress responses. However, scRNA-seq datasets compile data from thousands of cells and are characterized by high dimensionality, extreme sparsity, and considerable technical noise. In particular, most of the genes expressed in a specific cell are expressed in all cell types. Only a relatively small number of genes, so-called marker genes, are specific for each cell type. Assigning roles to individual cells therefore relies heavily on prior knowledge of the biological context and which genes are highly expressed in each cell type, making it difficult to identify marker genes and accurately assign cell types.
Against this background, a research team led by Dr. Qiang He and Dr. Aiguo Yang from the Chinese Academy of Agricultural Sciences developed PhytoCell, an ensemble learning framework for analyzing plant single-cell RNA sequence data (Figure 1). Their study was published online on March 27, 2026. crop journalaims to enable robust cellular biomarker identification and classification of cell subpopulations.
The research team’s PhytoCell framework integrates four machine learning models grouped as a computational stack. This “ensemble” of models leverages a powerful learning strategy that uses a portion of the data to identify biomarkers, which are then used to analyze the remaining data. Also, using four models instead of one model increases the predictive stability and generalizability of the framework. This framework ranks genes based on their importance to the overall data structure by calculating the maximum information coefficient and iteratively selects marker genes for model training.
To evaluate the performance of PhytoCell, the researchers used scRNA-seq datasets from corolla tissue. Nicotiana attenuate (coyote tobacco) were collected at three time points. They found that this framework was able to identify key marker genes and accurately classify cells into different cell states and subpopulations.
Next, to independently validate PhytoCell’s performance, the researchers turned to a large-scale scRNA-seq atlas generated in rice spanning multiple tissues, containing approximately 120,000 individual cells. Once again, the researchers found that the framework identified a set of biomarkers that unambiguously assigned cell states and grouped similar cells. Results showed that PhytoCell is robust and broadly applicable across plant species, effectively removing redundant noise, selecting core marker genes, and achieving accurate annotation of cell subpopulations.
Unlike traditional methods that rely on prior biological expertise, the team’s PhytoCell framework employs a purely data-driven strategy to identify marker genes, preserving the original structure of biological data even when using a minimal set of biomarker genes. The researchers found that in addition to several marker genes identified using traditional methods, the framework also identified additional genes that were clearly missed by other methods.
“These new biomarkers are valuable candidate genetic resources for crop improvement and cellular mechanism research,” said lead author Qiang He. “We have made PhytoCell available on a user-friendly web server for marker gene discovery and cell type annotation.”
This platform is available for free at https://cgris.net/phyto.
“Overall, PhytoCell provides a robust and scalable machine learning-powered solution for annotating cell subpopulations and identifying marker genes in plant scRNA-seq datasets,” said senior author and co-correspondent Aiguo Yang. “This further advances the integration of machine learning into plant genomics.”
###
Please contact the author:
Chang He
Email address: heqiang@caas.cn
Published by KeAi was founded by Elsevier and China Science Publishing & Media Limited to deliver high-quality research globally. In 2013, our focus shifted to open access publishing. We currently proudly publish over 200 world-class, open access, English-language journals across all scientific disciplines. Many of these are titles published by us in partnership with prestigious societies and academic institutions such as the National Natural Science Foundation of China (NSFC).
Research method
Computational simulation/modeling
Research theme
not applicable
Article title
PhytoCell: An ensemble learning framework for identifying cell states in plant scRNA-seq data
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Disclaimer: AAAS and EurekAlert! We are not responsible for the accuracy of news releases posted on EurekAlert! Use of Information by Contributing Institutions or via the EurekAlert System.

