It’s not just cells that are on their way to becoming skin pigment, blood, or nerves that undergo these changes. It responds to a dense web of molecular instructions, some advancing while others suppressing it. Biologists have become much better at tracking where cells are going. It has become much more difficult to identify which regulators are actually driving these choices.
That’s the problem a new model called RegVelo is trying to solve.
The framework, presented at bioRxiv, combines two areas of single-cell biology that have traditionally been treated separately: tracking how cells move during development and mapping the gene regulatory networks that shape that movement. RegVelo not only estimates the direction of likely changes in cells, but also attempts to identify the underlying interactions between the genes that cause those changes.
“For a long time, cell dynamics and gene regulation have mainly been modeled separately,” said Professor Fabian J. Theis, co-senior author of the study, director of the Center for Computer Health at Helmholtz München, and professor at the Technical University of Munich. “RegVelo brings these elements together, allowing us to examine not only how cells are changing, but also what regulatory interactions are helping to drive those changes.”
where the map ends
Single-cell tools are already providing researchers with a detailed view of development, depicted through Waddington’s landscape, where cells move along paths that diverge toward different identities. Two major approaches are central to that research.
One is pseudotime, which orders cells along their developmental path. The other is RNA velocity, which estimates the direction of change by comparing immature and processed RNA. These methods can tell you where cells are likely going, but they omit many of the control mechanisms that help send cells there.
At the same time, gene regulatory network methods are being used to infer which genes activate or repress other genes. Although these methods can identify possible wiring diagrams, they typically cannot predict how cells will move over time.
RegVelo was designed to connect these two views. Treating genes as members of networks rather than isolated units, regulatory factors influence target transcription in response to changes in cellular state. In practical terms, this means that this model aims to perform two tasks simultaneously: inferring developmental trajectories and simulating what happens when specific regulatory factors are changed.
This project was born out of collaboration between groups with different strengths. Tatiana Sauka Spengler, co-senior author and researcher at the Stowers Institute for Medical Research, contributed high-resolution gene regulatory circuits from her work on neural crest development. Theis’ group brought tools for RNA velocity and trajectory modeling. First author Weixu Wang is a postdoctoral fellow at the Computational Health Center and led the development of the complex deep learning system.
“What made this work particularly powerful was the combination of complementary strengths,” Sauka-Spengler said. “Our lab provided high-resolution gene regulatory circuits, and Fabian’s team, experts in the field, provided dynamic trajectory and network modeling. RegVelo was born by combining these two views into one framework for the first time.”
Testing the model on a mobile system
The researchers applied RegVelo to several biological systems, including the cell cycle, endocrine production in the pancreas, hematopoiesis, and neural crest development in zebrafish.
In cell cycle data from 1,146 U2OS-FUCCI cells, the model recovered the known direction of progression from G1 to S to G2M and produced a strong cross-boundary accuracy score of 0.864 out of 1. Its speed consistency reached 0.873, and the estimated latency showed a Spearman correlation of 0.683 with the protein-based FUCCI cell cycle score, which was used as the ground truth. agent.
The system also inferred regulatory relationships consistent with known biology. Among the most relevant factors were TGIF1 and ETV1, and their main targets included cell cycle genes such as: BUB1, TFDP1and TOP2A.
In pancreatic development, RegVelo restored all four final endocrine states (alpha, beta, delta, and epsilon cells). This analysis also suggested that some epsilon cells function as alpha cell progenitors, consistent with existing reports. When the team simulated perturbations in gene regulation, the model identified and highlighted known lineage factors. new rod 2 as a potential regulator of epsilon differentiation. Furthermore, it was pointed out that Neurod2-Rfx6 The interaction is particularly important for epsilon maturation.
The hematopoietic results were remarkable for another reason. Early RNA rate approaches were difficult in this system because hematopoiesis involves changes in transcription rates that violate the assumption of constant transcription. Although RegVelo makes no such assumption, it recovered all five terminal blood lineages in the dataset and accurately captured the known toggle-switch relationship between GATA1 and SPI1, a classical regulatory motif in red blood cell and monocyte fate decisions.
A closer look at neural crest determination
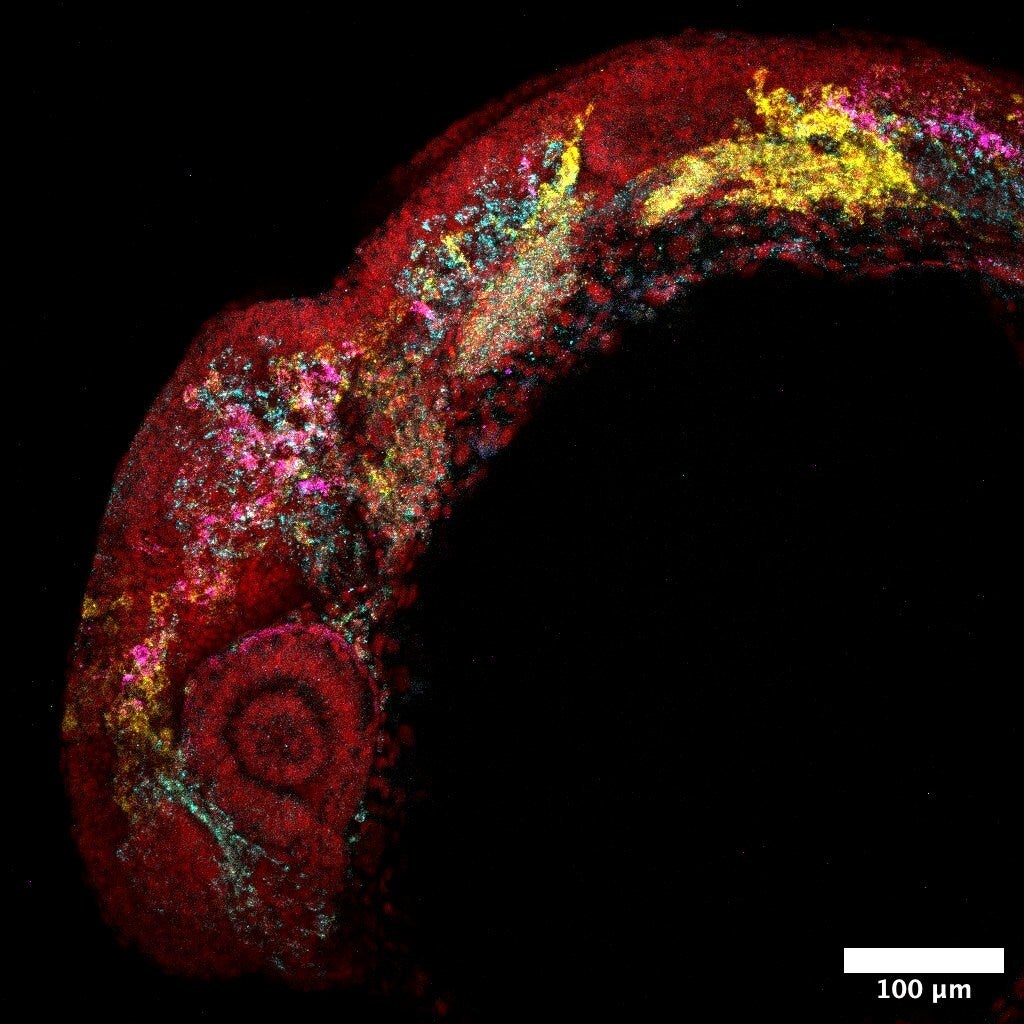
The study’s most detailed biological examination was obtained from zebrafish neural crest cells, an embryonic population that gives rise to pigment cells, peripheral nervous system elements, and craniofacial tissue.
Using Smart-seq3 data from 1,180 neural crest cells and derived cells across seven time points, the research team applied RegVelo in conjunction with a priori regulatory networks inferred from matched multi-ohm data. This model accurately restored the known terminal states of pigment cells, postauricular migratory neural crest, facial mesenchyme, and second pharyngeal arch cells.
From there, the researchers turned to predicting the perturbations.
RegVelo has been identified Tefek as an early driver of pigment cell development, ahead of other known pigment-related basic helix-loop-helix transcription factors. mitofa, Februaryand bhlhe40. Models are also ranked elf 1a transcription factor of the ETS family and one of the putative key regulators of pigment fate.
Both predictions held true in the experiment.
The research team used CRISPR/Cas9 knockout and direct capture Perturb-seq to Tefek Depleted pigment lineage. Perturb-seq also showed subsequent depletion of the pigment series. elf 1 On the other hand, hybrid chain reaction in situ hybridization revealed a decrease in pigment cells in both the cranial and postauricular regions of elf1 -deficient embryos.
This model did more than just name candidates. It suggested a regulatory background for them. For pigment lineages, RegVelo predicted: Tefek operate downstream of socks 10 and upstream of the propigmentation program include: elf 1. They also pointed out systems like toggle switches. elf 1 Promesenchymal ETS factors inhibit each other and help partition neural crest cells between pigmented and mesenchymal fates.
“Development is often described as a series of static snapshots of a cell’s state,” Sauka-Spengler says. “What we really want to understand is how cells make decisions and how they transition from one state to another. RegVelo models how these fate decisions are encoded into gene regulatory networks over time and what drives them.”
Wang said the framework allows him to test what happens if you remove one regulator. “We can derive testable predictions from single-cell data about which genetic regulators promote, slow down, or redirect particular developmental pathways,” he said.
Practical implications of the research
Although RegVelo is still a research tool, its appeal is clear. This gives scientists a way to move beyond descriptive cellular maps to models that can simulate how developmental pathways change when regulatory factors are perturbed.
This could help laboratories narrow down which experiments to run first, especially in systems where there are a large number of candidate regulators and limited testing time. It may also prove useful in disease settings where deregulated conditions lead to aberrant cellular states, such as developmental disorders, cancer, and regenerative medicine.
The findings also point to the broader idea of ”virtual cell” models that can predict behavior rather than simply cataloging it.
“RegVelo is a step toward virtual cell models that will help us better understand how cells behave in the context of differentiation and how they respond to genetic perturbations,” said Tice. “In the long term, this could help identify possible starting points for new treatments.”
Sauka-Spengler believes the experimental benefits could actually be achieved. “Having the full resolution of predicted, simulated, perturbed, and validated gene regulatory circuits gives us a very powerful tool,” she said. “We can start with stem cells or naive cells and develop new ways to coax them into cell types that can be used for cell therapy.”
The study results are available online in the journal bioRxiv.


