


Advances in deep learning have revolutionized the prediction of molecular structures, but real-world applications often require understanding not only single structures but also equilibrium distributions. Current methods such as molecular dynamics simulations are computationally intensive and insufficient to capture the full range of molecular flexibility. Prediction of equilibrium distribution is important for evaluating the macroscopic properties and functional state of molecules such as adenylate kinase. Deep learning shows promise for coarse-grained simulations, but struggles to generalize. Although the Boltzmann generator provides a potential solution by generating equilibrium distributions, its applicability between different molecules still needs improvement.
Researcher at Microsoft Research AI4Science in Beijing, China. University of Science and Technology of China, Microsoft Quantum, Redmond, Washington, USA.and Microsoft Research AI4Science (Berlin, Germany). Distribution grapher (DiG), a deep learning framework aimed at predicting the equilibrium distribution of molecular systems. Inspired by thermodynamic annealing, DiG uses neural networks to transform simple distributions toward equilibrium based on molecular descriptors such as chemical graphs and protein sequences. This allows us to efficiently generate diverse 3D structures and estimate the density of states much faster than traditional methods. DiG demonstrates versatility across different molecular tasks and can be generalized across different molecular systems. DiG approximates equilibrium distributions by simulating diffusion processes, facilitates prediction of molecular properties, and enables back-design of structures with desired properties.

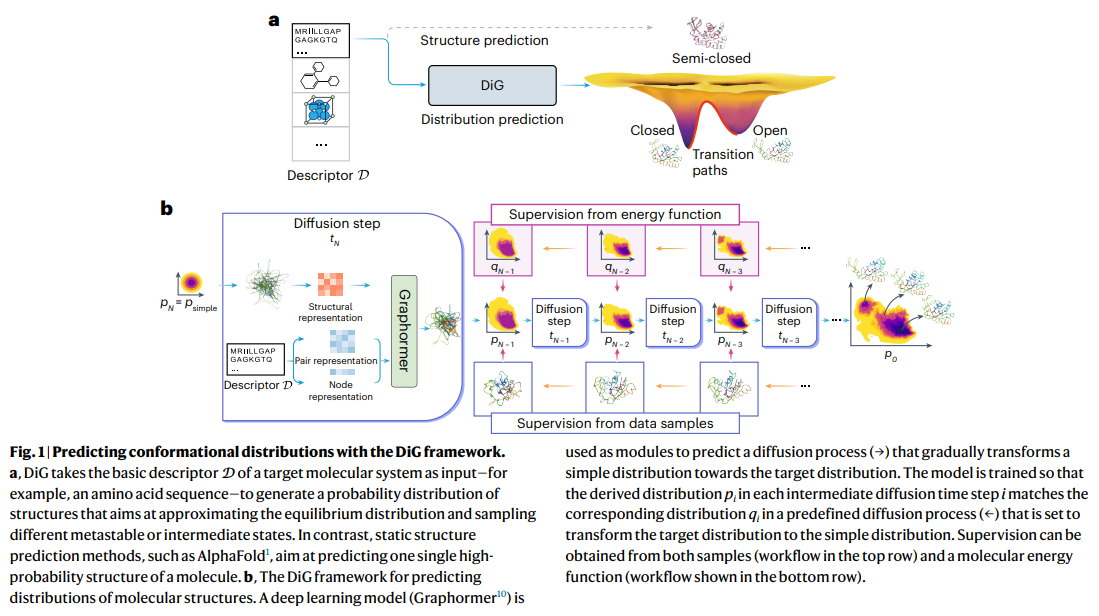
DiG, a deep learning framework, extends beyond the prediction of single molecule structures to the estimation of their equilibrium distributions. Inspired by the concept of thermal annealing, it uses a diffusion process to transform the target distribution into a simpler one, which is then reversed. Deep neural networks predict the inverse process by approximating a scoring function, facilitating the generation of diverse molecular structures. DiG also enables property-based structure generation and interpolation between states by mapping structures into latent space. This innovative approach advances molecular structure modeling, provides efficient prediction of equilibrium distributions, and facilitates property-based design.
DiG has demonstrated its versatility by successfully tackling a variety of molecular modeling and design challenges. Protein conformational sampling generates a variety of well-matched structures with energy landscapes that are important for understanding protein behavior and interactions. By leveraging experimental and simulation data along with innovative training methods such as PIDP, DiG accurately reproduces complex structural distributions, even for proteins with multiple functional states. Additionally, we demonstrate the ability to interpolate between states and provide insight into structural transition paths.
DiG provides extended range, excellent sampling of ligand structure around the binding site, and accurate prediction of ligand structure within druggable pockets. Its performance, validated against experimental data, highlights its potential application in drug design. Furthermore, DiG has demonstrated its ability in sampling catalyst adsorbate and efficiently identifying active adsorption sites on the catalyst surface. Its predictions are in close agreement with predictions obtained through computationally intensive methods such as density functional theory, and are notable for their speed and accuracy. Finally, DiG introduces the capability of property-based structure generation, enabling reverse design tasks such as carbon allotrope generation with desired electronic band gaps. This shows the potential to accelerate the materials discovery and design process.
In conclusion, DiG revolutionizes molecular science by efficiently predicting equilibrium distributions, enabling diverse molecular sampling important for understanding structure-function relationships and designing molecules and materials. Masu. DiG employs an advanced deep learning architecture to learn molecular representations from descriptors such as protein sequences and compound formulas to accurately capture complex distributions in high-dimensional space. The speed advantage compared to traditional methods such as MD simulation and MCMC sampling offers transformative potential and significantly reduces computational costs. With its ability to explore vast conformational space, DiG accelerates the discovery of molecular structures, impacting fields as diverse as life sciences, drug discovery, catalysis, and materials science.
Please check paper. All credit for this research goes to the researchers of this project.Don't forget to follow us twitter.Please join us telegram channel, Discord channeland linkedin groupsHmm.
If you like what we do, you'll love Newsletter..
Don't forget to join us 42,000+ ML subreddits


Sana Hassan, a consulting intern at Marktechpost and a dual degree student at IIT Madras, is passionate about applying technology and AI to address real-world challenges. With a keen interest in solving practical problems, he brings a new perspective to the intersection of AI and real-world solutions.

