SACs are composed of isolated metal atoms anchored to a support and coordinated by surrounding ligands. Since the concept was first proposed in 2011, SACs have been explored and utilized in various catalytic processes, spanning fields such as chemical engineering, energy, environmental science, agriculture, pharmaceuticals, and medicine1. SACs are particularly intriguing because of their ultrahigh atom utilization, atomically precise structures, and distinct properties compared to conventional NCs. To further expand their potential in real-world applications, there is a growing interest focus on the rational design of SACs116,117.
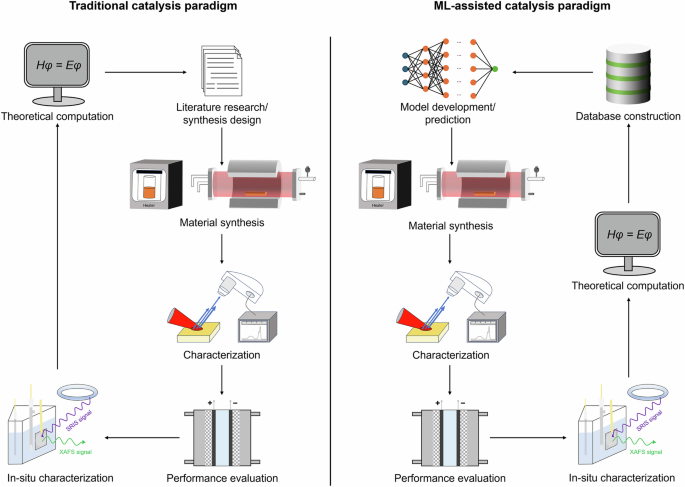
While empirical relationships (for example, volcano plots, scaling relationships, and Brønsted−Evans−Polanyi relations) and first-principle computations provide valuable theoretical insights for SAC design, their practical application is heavily constrained by lengthy computational time and high resource demands, as they require numerous calculations across a large parameter space. To address this challenge, the catalysis community has turned to data-driven ML as a fast, high-throughput, and computationally cost-effective tool to enhance SAC design118. Figure 6 illustrates how ML could aid SAC research, enabling deep analysis, the development of structure–performance relationships, and the discovery and prediction of desired catalysts. Thus, the ML-driven paradigm shift is revolutionizing the field of SACs.

Schematics of ML’s various applications in SAC design.
For example, ML has assisted researchers in successfully designing SACs with exceptional activity and selectivity by predicting adsorption energies (Eads) of key intermediates and Gibbs free energy change (ΔG) of elementary steps119. Additionally, ML can also be utilized in analyzing characterization data of SACs in a high-throughput manner, such as electron microscopic images and X-ray absorption spectra, to identify single-atom active sites responsible for catalyzing specific chemical reactions, therefore accelerating research greatly. The following subsection delves deeper into ML applications for SAC research, including the establishment of structure–performance relationships, high-throughput screening, and stability assessment of SACs.
Establishment of structure–performance relationship
Data-driven ML is a powerful tool for uncovering fundamental insights by establishing structure–performance relationships within large and complex datasets. In the design of SACs, ML can employ input feature engineering and feature importance analysis to identify the key factors influencing catalyst activity, selectivity, and stability in a specific reaction.
For instance, ML-driven DFT computations were adopted to explore the relationship between various structural properties of catalysts and hydrogen adsorption-free energy (ΔGH*) for HER. In detail, researchers built a variety of SACs consisting of N-doped carbon with different single-atom metals from the 3d, 4d, and 5d transition metals series, and computed their properties as input features. They then employed SISSO, a supervised ML algorithm, to accurately predict ΔGH* based on various features and compressed sensing, and identified key features for HER. Incorporated input features included d-state center (εd), covalent radius (rcov), Bader charge (q), number of occupied d states (docc), number of unoccupied d states (dunocc), Zunger radius (rd), number of valence electrons (Ne), ionization energy (IE), electronegativity (EN), and formation energy of single atom sites (Ef). The findings indicated that docc and q are the key features (the regression model achieved the highest accuracy using them as descriptors) in HER, while also deriving a fundamental descriptor for HER activity comprising four structural properties120.
For OER, multiple structural properties of SACs were used as input features to establish structure–performance relationships and predict the overpotential. A full connection NN (FCNN) model was trained on DFT-computed data, achieving high accuracy in predicting overpotentials (with a relative error of 6.49%) and significantly reducing computation time (Fig. 7a). It was found that the d-electron count (de), atomic radius of the metal (AtR), and electron affinity (EA) were the key features influencing the overpotential of OER. Additionally, an intrinsic descriptor (\(\phi\)) was introduced to quantify the overpotential of SACs based on their inherent structural properties with a combination of ML and DFT:
$$\phi ={{IE}}_{1}{{{\rm{d}}}}_{{{{\rm{e}}}}}{{{{\rm{At}}}}}_{{{{\rm{M}}}}}\left(\frac{{{EN}}_{{{M}}}}{{{At}}_{{{RM}}}}+\frac{{N}_{{{C}}}{{EN}}_{{{C}}}}{{{At}}_{{{RC}}}}\right)$$
where ENC, AtRC, and NC denote the electronegativity of carbon, the atomic radius of carbon, and the number of closest neighbor carbon atoms. ENM, AtM, and IE1 denote the electronegativity of the metal, the atomic mass of the metal, and the first ionization energy121.

a OER catalytic activities as an intrinsic descriptor of SACs on a single vacancy site and a double vacancy site. b Input feature–feature correlation map, including atomic, structural, and intermediate properties feature group. c Comparison between adsorption free energies of OH and descriptor φ‘OH for SACs embedded in Pyridine-4N and Pyrrole-4N local coordination, as well as the comparison between adsorption free energies of H and descriptor φ‘H for SACs embedded in Pyridine-4N. Reproduced with permission128. Copyright 2026, American Chemical Society.
Similarly, SACs’ structural properties like EN, EA, and AtR were utilized as input features to predict their ORR activity. First, comprehensive DFT computations were performed to generate data for ML. Researchers then created datasets from the DFT-computed data and identified potential input features using an integrated GBR algorithm. Predictive equations for ORR activity were subsequently proposed based on these key features. The method and findings of this research can be easily applied in the screening of other SACs, and greatly speed up the development of novel SACs for various purposes122.
In addition, structure–performance relationships for discovering and designing bifunctional SACs towards OER and ORR were established with GBR. The structural properties considered included TM bond lengths and coordination atoms (dTM-N1, dTM-C1, and dTM-C2), εd, charge transfer of TM atoms (Qe), EN, EA, the first IE (IE1), AtR, and de. The GBR predicted ΔG*OH with a high coefficient of determination (R² = 0.99) and a low RMSE (0.03 eV). Moreover, it should be noted that this study used only 16 data points, which is generally insufficient in most cases. Feature importance analysis revealed that IE1 and Qe are the most important features. IE1, which systematically increases horizontally across the periodic table, is a crucial factor influencing the activity of both OER and ORR123.
Furthermore, this methodology was applied to predict the catalytic performance of SACs in CO2RR. Using the GBR algorithm, one identified catalyst was Mo phthalocyanine with a proximal Ag atom, exhibiting a limiting potential of –0.33 V. Additionally, an intrinsic activity descriptor was proposed:
$$\phi ={E}_{1}{\theta }_{{d}}M\left(\frac{{E}_{{{M}}}}{{r}_{{{M}}}}+\frac{{N}_{{{C}}}{E}_{{{C}}}}{{r}_{{{C}}}}\right)$$
where EC and rC denote the electronegativity and atomic radius of carbon. NC represents the closest neighbor carbon atoms. This DFT–ML hybrid approach improved research efficiency by 6.87 times, with a mere prediction error of 0.02 V, paving the path for accelerating the rational design of advanced CO2RR SACs (Fig. 8a)124.

a Pearson correlation map between features, a feature importance map, and a heat map of ML-predicted theoretical limiting potentials of Pc dual-metal-site catalysts. b Volcano plots depicting relationships between the onset potentials for CO2RR, NRR, and ORR. Reproduced with permission124,131. Copyright 2026, American Chemical Society.
Structural properties such as de, oxide formation enthalpy (Hf,ox), the EN of the metal atom, the sum of EN of surrounding atoms, and the average pKa values of surrounding atoms were also incorporated as input features to establish structure–performance relationships. The RFR algorithm was employed along with DFT-computed data for 104 SACs embedded in graphene, encompassing M–C3, M–C4, M–pyridine N4, and M–pyrrole N4 configurations. This study identified de as the most important feature influencing ORR, OER, and HER activity of graphene-supported SACs. The developed RFR model was then used to predict the activities of 260 graphene-supported SACs (M–NxCy). Results showed that Fe–pyrrole N1C3 and Fe–pyrrole N2C2 exhibited higher activity compared to Fe–pyridine N1C3 and Fe–pyridine N2C2125.
For two-electron ORR, Guo et al. used a multiple linear regression method with eight structural properties as input features to analyze trends in the selectivity and activity of SACs. These properties included Hf,ox, the number of electrons in d/p orbitals (dpe), EA, EN, the number of coordinated nitrogen atoms (NN), the first ionization energy (IE1) of central atoms, the sum of electronegativity from neighboring carbon and nitrogen atoms (SEN), and the distance ratio (DR). Feature importance analysis identified Hf,ox and dpe as key factors influencing the ΔGO* of SACs. Metal centers like Ag, Au, and Pd, which have a lower oxygen affinity, were found to significantly reduce band hybridization between oxygen and the metal, thereby improving selectivity towards hydrogen peroxide126.
As the structures of SACs grow more complex, there is a pressing need for new and comprehensive descriptors to establish accurate structure–performance relationships. For instance, the number of isolated electrons in d-orbitals (Nie-d), based on a bidirectional activation mechanism, has been presented as a new descriptor for evaluating catalytic activities of SACs for NRR. The developed highly accurate SISSO model can greatly expedite the development of SACs127. Additionally, inspired by a feature importance analysis using SVR for porphyrin- and graphene-supported SACs, a linear dependent, elementary, and universal descriptor (φ) was proposed to describe the ΔG of OH*, O*, OOH*, H*, COOH*, CO*, and N2* for OER, ORR, HER, and CO2RR (Fig. 7b)128. In another work, using an extreme GBR model, Xu et al. modified the previously proposed descriptor φ, and proposed a modified descriptor φ‘, which incorporated the influences of the valence electron of the single-metal atom, the local coordination environment, and the intrinsic property L (the periodic number of the TM element in the periodic table) on the adsorption property:
$${\varphi }^{{{{\prime} }}}{{OH}}={\alpha }_{{{g}}}{\theta }_{{{d}}}\times \frac{{E}_{{{M}}}+\frac{1}{L-1}({n}_{{{N}}}\times {E}_{{{N}}}\times {n}_{{{C}}}\times {E}_{{{C}}})}{{E}_{{{O}}}}$$
$${\varphi }^{\prime} H={\alpha }_{g}{\theta }_{d}\times \frac{{{{{\rm{E}}}}}_{M}+\frac{1}{L-1}({n}_{N}\times {E}_{N}+{n}_{C}\times {E}_{C})}{{E}_{H}}$$
the EN of TM, C, N, O, and H atoms are represented by EM, EC, EN, EO, and EH, respectively. The coordination numbers of the first-neighbor N and C atoms of the metal center are nN and nC. A correlation coefficient αg is considered to take into account the slight reliance on the periodic table group of the element. The improved descriptor φ’ reflected the activity trends observed in studied SACs as well as facilitated the identification of SACs that could substitute noble-metal-based commercial catalysts. It was later proved that φ‘ is widely applicable for correlating SACs embedded in small, medium, and large macrocyclic complexes, provided that the active metal center’s local coordination environment doesn’t change (Fig. 7c)129.
Compared to SACs, DACs exhibit more intricate geometries, with the synergistic interaction between the two metal atoms being a significant factor influencing their performance. This complexity weakens the linear relationships observed in SACs, highlighting the need for advanced feature-engineering strategies and new descriptors that can capture these effects within the structure of DACs. Developing these features and descriptors is essential for accurately understanding and predicting the catalytic performance of DACs. Recently, an RFR-driven DFT was employed to construct the structure–performance relationships of DACs supported on nitrogen-doped graphene for ORR. This study revealed that the average distance between metal and nitrogen atoms (M12–N), the distance between metal atoms (M1–M2), and the outer electron quantity of metal atoms (Ne,O) are the key features regarding the limiting potentials for ORR130. Also, using a GBR model, Ren et al. developed a general and simple descriptor for designing 2D materials-supported DACs. The descriptor φ was presented as
$$\varphi =({\chi }_{{{{\rm{M}}}}}+\sum {\chi }_{X})+{N}_{{{{\rm{d}}}}/{{{\rm{p}}}}}$$
For a catalytic metal atom M interacting with a set of coordination atoms X, the terms (χM + ∑χX) and Nd/p represent the coordination environment, where \(\chi\) denotes EN and Nd/p represents the number of d or p electrons of the metal atom M. This descriptor effectively quantified the complex interfacial effects within the DAC systems, which governs the catalytic performance of the metal centers (Fig. 8b)131. Additionally, to identify general descriptors for DACs’ catalytic performance, Jia et al. systematically investigated the underlying structure–performance relationships. They discovered that the electronic and spectral descriptors, such as charge transfer, average metal charge, average d-orbital center on metals, and reactant stretching vibrational frequency, are good descriptors for O₂ binding132. Lin et al. introduced an interpretable descriptor model, ARSC, which decouples the atomic property (A), reactant (R), synergistic (S), and coordination effects (C) on the d-band shape of dual-atom catalysts (DACs). This descriptor significantly accelerates DAC design. To validate the model’s universality, Co2/NC and Ir1Co1/NC were identified as high-performance bifunctional electrocatalysts for both ORR and OER133.
To further understand structure–performance relationships, it is crucial to examine how various intermediates influence catalytic processes on SACs. Thus, besides structural characteristics, the properties of intermediates also needed to be incorporated as input features while training ML models. Fisher et al. categorized hundreds of topological features of SACs into three different feature groups: bond lengths and angles, statistical features, and partial radial distances. They employed these features to accurately predict the binding energies of *H, *OH, *O, and *OOH radicals on nitrogen-doped graphene SACs using random forest and SVM. Through their feature importance analysis, the type of intermediate was identified as the most influential feature134. In another research, Wang et al. employed the GBR algorithm to accurately predict the hydrogenation barriers for NRR. They found that the incorporation of intermediate features significantly improved the accuracy of the prediction, ultimately resulting in an impressive final RMSE value of 0.02 eV. The finding indicates a direct correlation between the structural features of intermediates and their ΔG135. Furthermore, a descriptor-based design was proposed to develop active SACs for CO2RR by establishing a correlation between catalyst activity and the ΔG of two intermediate species (*OH and *OCH). This approach revealed that Ni, Cu, and Co are effective metal centers for SACs in CO2RR136.
The descriptor could also be utilized to establish volcano-shaped relationships, facilitating the identification of SAC candidates suitable for various catalytic reactions. Gong et al. introduced a novel descriptor based on the bonding, topology, and electronic structures of the metal centers of SACs, which correlates with catalyst activity:
$$\phi =\frac{{N}_{{{{\rm{e}}}}}{EN}}{{I}_{{{{\rm{R}}}}}}$$
where Ne and IR represent the valence electron number and ionic radius of the central metals. This descriptor was employed to generate volcano plots for overpotential, onset potential, and Faraday efficiency, and showed two distinct peaks in the overpotential plot, with Ti and Co positioned at the summits137.
In another study, 9 classification and 15 regression algorithms were used to predict the energy barriers of C–H dissociation across various single-atom alloys (SAAs). SAAs are an important subclass of SACs. Unlike supported SACs, alloy environments offer fewer coordination motifs and are less prone to restructuring under reaction conditions, leading to more stable and predictable reactivity and selectivity138. Based on these predictions, Ir1Ni and Re1Ni were identified from a library of 10,950 samples as top performers for methane cracking. Notably, Re1Ni achieved an H2 yield of 10.7 gH2 gcat–1 h–1 with 99% selectivity and 7.75% CH4 conversion at 450 °C (Fig. 9a)139. Lin et al. employed symbolic regression and compressed sensing to identify the key features determining NRR activity. They introduced a simple intrinsic descriptor and used an SISSO model for feature importance evaluation and descriptor training, which effectively accelerates the high-throughput screening of electrocatalysts based on the constructed structure−activity relationship. An experimental volcano plot including 13 previous reports and their synthesized four materials was plotted to validate its feasibility. One of the materials involved showed the highest activity (Ru−N3), which is in good agreement with the descriptor’s guidance (Fig. 9b−d)140. Moreover, five ML algorithms, including linear regression, RFR, GBR, SVR, and KRR, were used to identify the optimal descriptor for analyzing how various physical and chemical properties of metal atoms influence the adsorption or reaction energy of the metal with sulfur, Na2S, and Na2S4. Accordingly, a synergistic interaction between the adsorption model and electronic transfer was established. It was found that the charge-transfer process facilitates the rearrangement of sodium ions, ultimately improving pathway selectivity and conversion to stable products during the redox process, thereby enhancing the electrochemical performance of room temperature sodium–sulfur batteries141.

a Total C–H dissociation rate on all surfaces of the ML-designed SAA catalysts at 450 °C. b Relationship between –UL versus the descriptor \(\phi\)0.5 × \(\bar{\chi }\)–1.5 on TM-NC SACs. c Volcano plot for –UL versus the descriptor \(\phi\)0.5 × \(\bar{\chi }\)–1.5 on central metals with other nonmetal-doped coordination environments in the range 0.70–0.82. d Experimental NH3 production rates for NRR versus the descriptor \(\phi\)0.5 × \(\bar{\chi }\)–1.5 of both previously reported and the synthesized Ru/Mo–NC–T materials. Reproduced with permission140. Copyright 2026, John Wiley and Sons.
In ML-assisted SAC design, descriptor selection should be tailored to the target reaction, data availability, and model characteristics, which together govern model accuracy and interpretability. For example, atomic and electronic descriptors (including d-electron count, electronegativity, ionization energy, and charge transfer) effectively describe the intrinsic properties of single-atom sites and are well suited to interpretable models and small datasets, whereas geometric descriptors (including coordination number and bond length) are crucial for capturing local environments and metal–support interactions. For reactions involving multiple intermediates, incorporating intermediate-related descriptors can markedly improve predictive accuracy. When descriptors are not well matched to the problem or model complexity, reselection or modification of the input features should be considered. Despite this, there remains a lack of universal and suitable descriptors for the myriad of SACs, supports, and catalytic reactions. Such a challenge lies in the highly localized electronic structures of single-atom catalysts, as well as their dependence on and sensitivity to metal–support interactions. In addition, different reactions may involve entirely distinct mechanisms, severely limiting the transferability of descriptors. Consequently, substantial amounts of both computational and experimental data are still required to train ML models, optimize feature-selection strategies, and refine the employed ML algorithms. This approach allows for the development of more effective descriptors for SAC design.
High-throughput computational screening
DFT computation has seen wide application in the high-throughput screening of SACs. However, the application of DFT is often constrained by its high demand for computational resources. ML, with its data-driven nature and strong generalization ability, offers a promising solution to this limitation. ML can significantly reduce time and effort by identifying similarities among various SACs and accurately establishing structure–performance relationships, thereby accelerating the screening process. As a result, researchers have increasingly integrated ML algorithms with DFT computations to enhance the high-throughput screening of SACs.
For instance, ML-integrated DFT calculations have been used for the screening and designing of SACs supported on two-dimensional metal borides (MBenes) for HER. The SVM-based ML model accurately calculated the ΔGH* values, and the Bader charge transfer of the surface metal was identified as the key feature influencing HER activity. Among the candidates, Mn supported on Co2B2 was found to be a highly efficient HER catalyst, as its |ΔGH*| values were <0.15 eV142. Similarly, a hybrid DFT–ML approach was utilized to facilitate the rational design of high-performance SACs supported on 2D materials for the HER. 364 SAC models were systematically designed by embedding 3d, 4d, and 5d single-metal atoms into various supports, encompassing g-C3N4, π-conjugated polymers, pyridinic graphene, and hexagonal boron nitride. An SISSO model was conducted on multiple electronic, geometric, and thermodynamic descriptors, enabling the identification of stable and high-performance SACs. Notably, SACs such as Pd–B4, Ru–N2C2, Pt–B2N2, Fe–N3, Fe–P3, Mn–P4, and Fe–P4 exhibit near thermo-neutral binding energies (|ΔGH* | = 0.01–0.02 eV), indicating their excellent HER activities143. Jyothirmal et al. conducted a comprehensive study combining DFT computations and ML to identify suitable single atoms for anchoring on g-C3N4. By screening a wide range of elements based on their formation energies, they identified B, Mn, and Co atoms supported on g-C3N4 as promising catalysts for hydrogen production. Further analysis, using SVR coupled with feature engineering, highlighted that formation energy, bond length, boiling point, melting point, and valence electron configuration are the most influential factors of the SACs’ HER activities144.
Using ML, researchers efficiently evaluated the stability and catalytic activity of 3d, 4d, and 5d TMs for ORR. The subgroup discovery-based ML model, trained on carefully selected features, revealed that the most active structures possess a medium d-band center and Bader charge, which directly affect the adsorption strength of key intermediates and, therefore, the catalytic performance. The study also found that the stability of these materials is determined by a complex combination of EN, the number of outer electrons of the metal center, the d-band center of metal oxides, and the relative coordination number of adsorbed species (Fig. 10a)145. Additionally, Sun et al. developed a geometric and electronic informed overpotential model (GEIOM) using a random forest algorithm to perform high-throughput screening of candidate SACs for ORR. The ML model demonstrated remarkable accuracy, achieving an R2 of 0.96 and an RMSE of 0.21. From this screening, 30 promising catalysts were identified, exhibiting a low RMSE of 0.12 V (Fig. 10b)146. Wang et al. conducted a comprehensive DFT study on 48 SACs comprising eight classic carbon-based substrates (graphdiyne (GDY), C2N, C3N4, phthalocyanine, C-coordination graphene, N-coordination graphene, covalent organic frameworks and metal-organic frameworks) and 3d, 4d, and 5d TM elements (Cr, Mn, Fe, Co, Ni, and Cu). The generalized gradient approximation with the Perdew–Burke–Ernzerhof functional was used to describe the electron exchange-correlation interactions. Among the 48 SACs containing six metal atoms and eight carbon supports, Co1/MOF, Ir phthalocyanine (IrPc), Co–N–C, Co1/GDY, and RhPc were identified as promising catalysts for ORR due to their predicted low overpotential (~0.35 V) and high stability147. In another study, an SISSO-based screening strategy was proposed to efficiently identify catalysts with superb selectivity and activity for the two-electron ORR for H2O2 production. By analyzing the relative stability of reaction intermediates and their underlying mechanisms, this approach significantly accelerated the screening process. A database of 150 N-doped graphene-supported SACs was constructed and evaluated. Using ΔGO and ΔGOOH as key descriptors, the most promising SACs, such as PdN4, PtN4, NiN4, and CuN4, were rapidly identified for the two-electron ORR148.

a The anchor energy map based on high-throughput screening results with five adsorption intermediates (*COOH, *OCHO, *CO, *CHO, and *H) for CO2RR. b Optimized configurations with high activities and stability for ORR. Reproduced with permission145,146. Copyright 2026, American Chemical Society.
DFT computations combined with ML were also used to screen potential MXene-supported SACs for OER by assessing their stability and cohesive energy. Ti3C2(OH)x, V3C2(OH)x, Zr3C2(OH)x, Nb3C2(OH)x, Hf3C2(OH)x, Ta3C2(OH)x, and W3C2(OH)x were identified as highly stable candidates. Zn, Pd, Ag, Cd, Au, and Hg were identified as promising single atoms to be anchored on MXenes due to their high cohesive energy. Based on the combined results, Hf3C2(OH)x with a Pd single atom showed a theoretical overpotential of 81 mV for OER149. Similarly, researchers developed an FCNN model in PyTorch and optimized it using DFT-computed data to predict OER overpotential. The model demonstrated impressive accuracy, with mean relative errors of 6.70% (training data) and 6.49% (testing data), while significantly reducing the computation time compared to manual DFT calculations119.
ML-integrated DFT methods have also been employed to rapidly screen efficient catalysts for NRR and CO2RR. For instance, a graph-based CNN was utilized to expedite the screening of SACs for NRR, revealing that the N–N bond length significantly impacts catalyst activity, as well as the importance of N2 activation for achieving high catalytic performance150. In another study, a DNN was utilized for fast and high-throughput screening of high-performance SACs embedded on boron-doped graphene for NRR. Adsorption and free energies of intermediates were calculated with a light gradient boosting machine (LGBM) model. Feature importance analysis further identified the coordination number of the metal center and the number of hydrogen atoms as critical features influencing catalytic performance151. In the context of CO2RR, Chen et al. employed extreme GBR to screen ΔGCO* and ΔGH* across 1060 SACs supported on graphene, using simple structural features. Feature importance analysis identified the EN, rcov, and IE1 of the metal atoms as the most critical features influencing ΔGCO*152. Furthermore, Xi et al. conducted a comprehensive high-throughput screening of 192 SACs supported on monolayer TM dichalcogenides to investigate the correlation between their intrinsic properties and catalytic activities for CO2RR. The study utilized SISSO to identify key descriptors that influence the performance of SACs. Among the screened catalysts, Fe1/CoS2, Pt1/TiTe2, and Co1/CoS2 exhibited outstanding performance, characterized by low limiting potentials for CO2RR and highly selective pathways towards various products. Notably, the study revealed a robust linear relationship between the difference in covalent and atomic radii of the metal center and the charge transfer of *COOH, highlighting its critical role in multiple reaction steps153.
ML can enhance not only computational processes but also experimental workflows. For example, Mitchell et al. developed a customized deep-learning method for automated atom detection in image analysis, a crucial step toward high-throughput TEM. The model identified over 20,000 atomic positions for statistical analysis of various reactions, significantly accelerating image processing and reducing human bias by providing an uncertainty analysis, which is difficult to achieve through manual atom identification. As a result, the standardization of experiments was achieved, and scalability was greatly improved (Fig. 11a–d)154. Moreover, Martini et al. demonstrated that combining supervised and unsupervised ML approaches could effectively decipher the X-ray absorption near-edge structure (XANES) of M–N–C materials. Their results revealed that the single-atom Ni sites are the active species for CO2RR, highlighting their dynamic, complex nature and adaptability to the reaction environment (Fig. 11e–h)155. Similarly, Zhao et al. conducted a LASSO-driven analysis of IR spectroscopy to deduce the pyrolysis process of Pt-doped zeolitic imidazolate framework-67 (ZIF-67) for synthesizing Pt1/Co3O4 SAC. The algorithm provided correlation coefficients for the selected structures, confirming crucial structural changes over time and temperature, and revealing the formation mechanism of SACs. The study also demonstrated that the integrated approach—combining ML algorithms, theoretical simulations, and experimental spectral analysis—can effectively interpret experimental characterization data and shows great potential for broader applications (Fig. 11i–l)156.

a Typical background-subtracted and intensity-normalized STEM image of the Pt1/NC catalyst. b Likelihood map for atom detection generated by the SAC-CNN model corresponding to (a). c Processed STEM image with the manually tagged (red circles) and SAC-CNN-detected (yellow crosses, yellow circles) single atoms overlaid. d SAC-CNN-generated likelihood landscape. e–g Comparison of the XANES component 1 (e), component 2 (f), and component 3 for pure species, as extracted from the experimental data, with the best-fit results (g). h Points in a structural parameter space obtained using the adaptive sampling. i–l Experimental characterization outcomes (i, k) and ML-simulated in situ temperature-dependent DRIFTS spectra for the pyrolysis processes of ZIF-67 and Pt-doped ZIF-67 (j, l). Reproduced with permission154,155,156. Copyright 2026, American Chemical Society.
Stability prediction
High stability is another crucial requirement for developing high-performance SACs, which necessitates investigating metal–support interactions, aggregation energies, and structural changes caused by adsorbates. To achieve strong metal–support interactions in SACs, it is recommended to form robust coordination bonds around the metal center. This can be achieved by regulating the metal–ligand bonding. Common strategies include selecting different coordinating elements in the first or second coordination shell, adjusting the coordination number, controlling bond lengths, and fine-tuning bond angles. These factors influence the strength and stability of the metal–ligand bond, as well as the electronic and geometric structures of SACs.
In this context, ML could work as a powerful tool to assist researchers in designing and synthesizing SACs with high metal loading and strong metal–support interactions. For instance, ML-driven DFT computations have been used to establish correlations between the stability of SACs on oxide substrates and parameters such as binding energy (Ebind) and the cohesive energy of the bulk metal (Ec). Using three different algorithms, including ridge, LASSO, and elastic net regression, a close relationship was found between the diffusion activation barrier (Ea) and Ebind2/Ec within the space of physical descriptors. This finding differs from previous results that linked Ebind directly with Ec. This diffusion scaling law offers a simple model for evaluating the thermodynamics and kinetics of supported metal atoms, thereby accelerating the design of advanced SACs with robust metal–support interactions (Fig. 12a)157,158.

a Lifetime of SACs estimated by the characteristic time of diffusion depending on the binding energy of single-metal atoms (Ebind) and the cohesive energy of the metal (Ec). b Stability map of single-atom alloys relative to configurations. Squares with different colors represent different stability energies compared to the most stable configurations (Dark blue squares). Reproduced with permission157,158,160. Copyright 2026, American Chemical Society.
To ensure that the designed SACs exhibit high thermodynamic stability, researchers have also utilized ML to study the tendency of atoms to diffuse into the bulk material, aggregate into surface clusters, and avoid alloying with the host. Rao et al. studied the stability of several SAA configurations, creating a 28 × 28 database by selecting 26 d-block metals and combining them with Al and Pb, resulting in 250 highly stable combinations. Additionally, another database was constructed, comprising 358 systems where the SAA geometry is within a small energy difference (0.5 eV) from the ideal configurations. Decision trees, neural networks, and SVM (for classification of data) using structural properties as input features were employed to classify the DFT-computed data. Furthermore, a physical bond counting model was integrated with a KRR algorithm to broaden the model’s applicability, allowing it to be extended to similar geometries excluded in the training data159. The thermodynamic stability of SAAs was further assessed by evaluating aggregation energies and structural changes induced by adsorbates such as *O. The employed algorithms were trained on DFT-computed data for 38 different copper-supported SAAs. A GPR model was used to predict aggregation energy and *O adsorption energy, achieving MAE of 0.092 and 0.091 eV, respectively. The GPR model’s versatility also enables its application to various supports, different adsorbates, and larger cluster sizes, addressing the numerous degrees of freedom involved and contributing to a reduction in computation time (Fig. 12b)160.
The stability of GDY-supported SACs, in terms of their zero-valence state and electron transfer capabilities, was assessed using a combination of ML and DFT. The analysis revealed that among TMs, zero-valence Co, Pd, and Pt SACs exhibit high stability, as indicated by the energy barrier differences between electron gain and loss. The unsupervised ML algorithm Fuzzy C-Means was employed to categorize the DFT-computed data. This ML approach was used to construct a database to aid in the screening of SACs embedded in GDY. The researchers also examined the effects of varying the number and directions of electron transfers between the metal center atoms and GDY, identifying the starting single-electron transfer as the most unfavorable one161.
The stability and activity of SACs embedded in NxCy were evaluated for HER, OER, and ORR with descriptors derived from DFT and ML. Among the varied active-site configurations in SACs, M1N2C2 was found to exhibit superior electrochemical catalytic performance, easier formation, and enhanced durability without aggregation or dissolution. Using M1N2C2 as templates, Ni, Ru, Rh, and Pt were identified as having low overpotentials for HER. For the first time, it was demonstrated that both metal center atoms and carbon atoms are involved in H adsorption. The results highlighted the important role of coordination environments in achieving high stability and guiding the design of high-performance SACs162.
Common features and descriptors for ML models in SAC design
SVM, KRR, RFR, and DNN are the primary supervised ML algorithms employed to elucidate the correlation between input features and SAC performance. These algorithms are typically implemented using Scikit-learn. Structural properties, like the number of electrons in the d orbital, are commonly used as input features. While ML has shown great potential, its broader applicability remains limited by the inconsistency of available experimental and computational datasets and by the strong system- and task-specificity of existing models, which hampers transferability across catalytic systems.
Furthermore, descriptors like the d-band center, Bader charge, IE, EA, rcov, the number of electrons in the d orbital, formation energy, and oxide formation enthalpy are commonly used to describe the catalytic activity of SACs. A major challenge in ML’s application to SAC research is the limited availability of suitable descriptors to use as input features. An ideal descriptor should meet the following criteria: physical interpretability, relative simplicity, and significant feature importance in ML models. However, ML techniques can sometimes obscure the physical interpretation of descriptors like the d-band center and enthalpy of vaporization, making it more difficult to understand their effects on catalytic properties.
In addition, data accessibility is another important factor when considering an ideal descriptor. For instance, descriptors like frontier molecular orbitals and the density of states (DOS) may be suitable, but they require extensive DFT calculations, which limit their practicality. On the other hand, descriptors based on the properties of metal atoms and supports, like atomic number, number of d-orbital electrons, IE, and coordination number of metal atoms, offer convenience and can be easily obtained without the need for time-consuming DFT computations.


