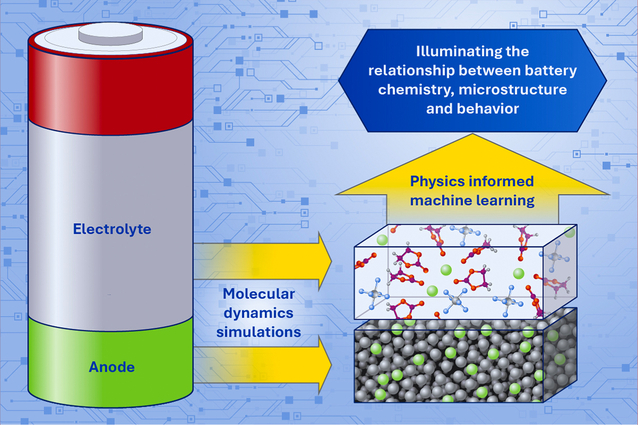
America needs better batteries for grid-scale energy storage and national energy resiliency. Scientists at Lawrence Livermore National Laboratory (LLNL) are tackling this challenge in a variety of ways, but one approach that’s making a big impact is physics-based machine learning.
In two recent publications, LLNL researchers examined how integrating molecular dynamics simulations and physics-based machine learning can uncover structure-behavior relationships in complex battery materials. They used a powerful combination of techniques to study carbon anodes in sodium-ion batteries and liquid electrolytes in lithium-ion batteries.
“These studies demonstrate that the structural complexity of battery materials is not only a hindrance to understanding, but also a design advantage, and lays the foundation for high-throughput screening of next-generation energy storage materials,” said LLNL scientist and author Liwen (Sabrina) Wang. “By encoding that complexity into physics-based machine learning models, we can predict properties and identify design levers that are inaccessible with traditional approaches.”
The first paper, published in Energy Storage Materials, investigated sodium-ion batteries. Because sodium is abundant and available domestically, this technology is critical to ensuring a strong U.S. supply chain.
Sodium batteries work by shuttling sodium ions from the anode to the cathode. The most commercially mature sodium anodes are made of hard carbon, which looks like a jumble of crumpled, disordered graphene-like sheets. This disordered structure, with many small pores and empty spaces, makes it difficult to characterize and design the anode.
“Sodium ions can move through all of that disorder, slipping between layers, depositing on surfaces, and filling nanopores,” said LLNL scientist and author Nikhil Rampal. “That complexity is part of what makes hard carbon so promising, but it’s also what makes it so difficult to design.”
Researchers have long struggled to understand how the characteristics of atoms within hard carbon relate to the transport of sodium ions. In this study, the team used LLNL’s high-performance computing to simulate how all the atoms in the material move and interact over time.
“We essentially created an atom-by-atom video of sodium ions being diffused, clustered, or trapped within the carbon,” Rampal said.
The authors then used these movies to train a machine learning algorithm to predict how the atoms would interact. This algorithm can run much larger, longer, and more accurate simulations at an affordable price. This was used to classify the movement of sodium ions into eight different regions based on their unique interactions with hard carbon.
“Increasing carbon density and sodium loading causes ions to cluster or become trapped within the nanopores, which has a direct impact on rate capability and thermal safety,” Rampal said.
The result is a quantitative map between microstructure and ion transport, including a practical way to strengthen hard carbon. The researchers believe this work provides a concrete path to safely maximize sodium ion transfer and, in turn, enable the deployment of sodium battery technology.
A second paper published in EES Batteries applies the same philosophy to a different challenge: better electrolytes for lithium-ion batteries. Designing the ideal electrolyte is a combinatorial challenge, as the infinite possibilities of solvents, salts, additives, and concentrations are too vast to exhaustively screen.
Traditional electrolyte models rely on text-based representations that ignore the 3D geometry of molecules. In contrast, the LLNL team used molecular dynamics simulations to generate realistic 3D configurations of molecules. They fed these structures into a machine learning model that predicted the statistical stability of each configuration.
The key insight is that electrochemical stability depends on the assembly of the whole molecule, not just the sum of its parts.
“The identity and concentration of the salt or solvent can dramatically shift the predicted stability window through mechanisms that are simply not visible to text-based encoders,” Rampal said. “For example, exchanging one lithium salt for another widened the stability range by 57%. This is determined entirely by how the anions are arranged around the lithium ion.”
Scientists envision this molecular dynamics and physics-based machine learning pipeline as a high-throughput screening platform that replaces trial-and-error electrolyte design with physics-based exploration. Incorporating experimental benchmark data improves model accuracy over time and allows the core principles to naturally transfer to other battery chemistries and electrochemical systems.
“This could significantly accelerate discovery for national laboratory programs exploring large design spaces across lithium, sodium, and multivalent battery chemistries,” Wang said. “Although these studies focus on batteries, the broader framework can be applied to many other systems.”
Authors on LLNL’s hard carbon research also include Stephen Weitzner, Marissa Wood, and Jonathan RI Lee. Funding support is provided in part by LLNL’s Institute-Directed Research and Development Program and in part by the U.S. Department of Energy (DOE), Office of Power, Division of Energy Storage. Computing support was provided by the LLNL Institutional Computing Grand Challenge program, with resources sponsored by the DOE Office of Critical Minerals and Energy Innovation and located at the National Laboratory of the Rockies.
/Open to the public. This material from the original organization/author may be of a contemporary nature and has been edited for clarity, style, and length. Mirage.News does not take any institutional position or position, and all views, positions, and conclusions expressed herein are those of the authors alone. Read the full text here.


