Companies at the forefront of aerospace, energy, and computing are constantly searching for new materials to improve performance. But to understand how those materials actually work inside a rocket or on a computer chip, companies must first manufacture them and then test them. That’s because even the most powerful simulation techniques have difficulty modeling the complex chemical configurations in most of today’s solid-state materials. This problem adds cost and time to materials innovation.
Now, a team of MIT researchers has developed a method to accurately model the behavior of metals, regardless of the complexity of their chemical configuration. At the heart of this approach is a machine learning model that makes material simulation faster and more accurate. The researchers improved these models by building training datasets that capture the diversity of atomic environments in chemically disordered materials.
in a new paper scientific progressthe researchers showed that their approach could be used to accurately predict the material properties of different groups of metal alloys under different conditions. They also showed how this approach can be used to develop new materials, especially in scenarios where experimentation is expensive.
“Although the focus of this paper is metal alloys, which is my area of expertise, this could potentially apply to other types of materials, such as semiconductors,” said lead author Rodrigo Freitas, TDK Career Development Professor in Materials Science and Engineering at MIT. “This is not application-specific. We can use this approach to create things like new sustainable steels and new materials for aerospace. That’s what makes this exciting.”
Joining Freitas on the paper is lead author Dr. Killian Sheriff ’26. MIT doctoral students Daniel Xiao and Yifan Cao. and Lewis R. Owen, Senior Lecturer at the University of Sheffield.
metal modeling
The properties of materials are primarily determined by the internal arrangement of chemical elements. Even if two materials have the same combination of chemical elements, differences in chemical arrangement can make the difference between a material that is brittle and one that deforms without breaking.
To understand this difference, we need to simulate the material atom by atom. To do that, researchers rely on models that explain how atoms interact. Over the past two decades, machine learning has become the most accurate way to build these models. Such models work well when the chemical arrangement inside the material follows a highly ordered pattern, but this is not the case for most solid materials, where the chemical arrangement of atoms is disordered and varies from region to region.
“The real challenge in our field is to model these chemically disordered phases,” Freitas says. “Chemical disorder means that the local chemical environment is very diverse, which is difficult for machine learning models to learn. This is a problem because all the metals we use in practice are chemically disordered.”
The problem ultimately lies in the lack of representative training data for atom-by-atom simulations. Current leading approaches to creating such data work in a brute-force manner, often requiring more than 100,000 hours of computation to create training data for a single material. Still, when researchers change the material’s composition, it doesn’t transfer well.
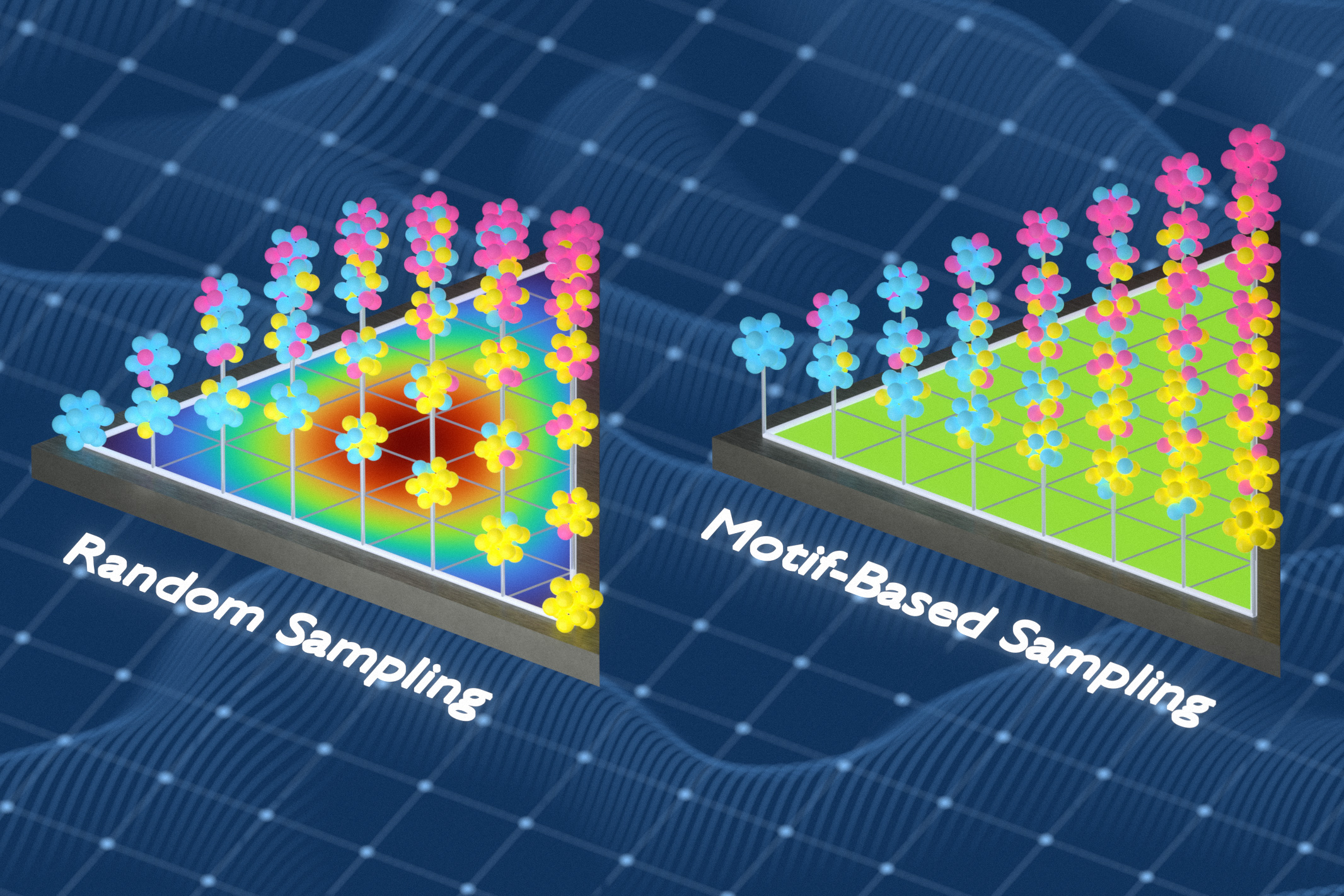
In previous work, Freitas’ group developed a method to measure the chemical complexity of solid materials by analyzing the frequency and spacing of small atomic groups. In this study, the researchers used that feature to build a better training dataset. They used a mathematical approach known as information theory to generate a training dataset that captures different local chemical environments inside the disordered material. This method works by replacing atoms from the sample, reducing repetition and exposing the model to a chemical environment that could otherwise be missed.
“We continued to optimize our training set to capture as many different local environments as possible,” Freitas says. “When the same type of environment appeared over and over again, we replaced the redundant examples with examples the model had never seen before. This made the training set more informative because each example added something new.”
Models trained on the researchers’ dataset predicted material properties more accurately than models trained using random sampling or other common sampling methods.
“The starting point for all these atom-by-atom simulations is whether we can accurately describe the chemical bonds between atoms,” Freitas explains. “Even if you don’t, you can learn about materials in general, but you don’t know what will happen to a particular material in the real world. This approach allows the simulation to have higher fidelity from a chemical standpoint and better reflect what is happening to the material.”
The researchers applied their technique to create a machine learning training dataset for a chemically diverse group of metal alloys. Using a series of machine learning models, they showed that models trained on their own datasets were more accurate than much larger models created by companies like Google and Microsoft.
“We’ve gotten to the point where we believe it can work without using these expensive brute-force methods,” Freitas says. “I said to Killian, “This is a good paper, but if you can show that simulations using these models can accurately predict useful material properties, that’s a very good paper.” Killian took that to heart and tested it as extensively as possible.
Sheriff worked with Xiao and Kao to test approaches across different alloys and properties. The research team also used Owen’s experimental data to compare their simulations with actual measurements of atomic order within the alloy.
From the lab to industry
This method works, in part, by capturing hidden patterns in the sample data. The researchers describe the patterns in their paper as “subtle energy biases for specific local chemical configurations.”
These small energy differences are important because they determine which phases form within the alloy, how those phases change with temperature and composition, and ultimately what properties the material has. As one test, Daniel Hsiao led a simulation that showed the team’s model could predict a phase diagram that closely matched experimental data. Phase diagrams map which phases are stable over different temperatures and chemical compositions and are a central tool for alloy design and processing.
“Phase diagrams are one of the main ways to connect material modeling to actual processing decisions,” says Freitas. “If you’re welding, casting, or heat treating alloys, you need to know which phases are likely to form under different conditions. Our goal is to make these kinds of predictions accurate enough and accessible enough that they become part of how people design materials.”
Researchers are currently using this approach to study how changes in alloy composition affect mechanical properties and radiation resistance, with the goal of designing materials that maintain strength and damage resistance in harsh environments. They are also working to make the method easier to use with the kinds of tools and workflows materials engineers already utilize.
“If what you’re building doesn’t fit with existing operating procedures, the industry isn’t going to change the way it does things,” Freitas says. “The goal is to make these predictions useful in the real world, where important decisions are made.”
This research was supported by the U.S. Air Force Office of Scientific Research.