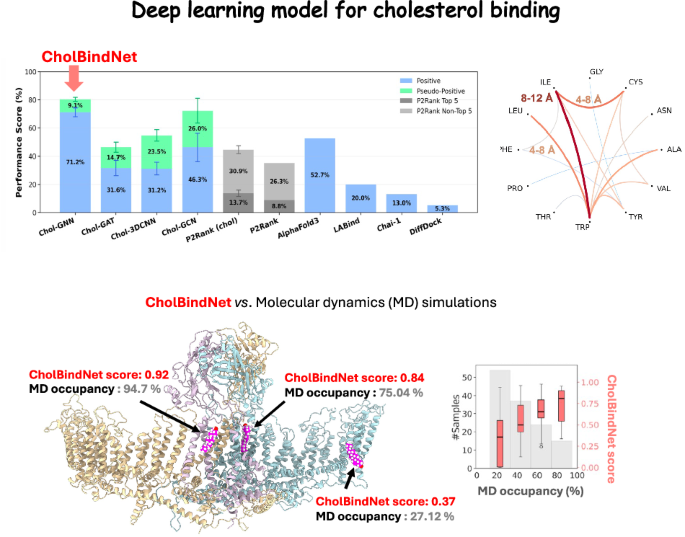
Although cholesterol is an important regulator of membrane protein structure and function, predicting cholesterol binding sites remains difficult due to its non-drug physicochemical properties. Here, we selected more than 800 high-resolution transmembrane protein structures, including cholesterol, and developed an interpretable atom-based graph neural network called CholBindNet. To address the lack of negative samples due to the promiscuous nature of cholesterol binding, a positive unlabeled (PU) training strategy was adopted. We show that CholBindNet significantly outperforms existing machine learning models trained on popular ligand binding datasets such as AlphaFold3, P2Rank, and DiffDock. The performance and generalizability of the model to invisible membrane proteins was further demonstrated by rapidly evaluating the cholesterol binding site of the PIEZO2 ion channel against all-atom molecular dynamics (MD) simulations performed on the Anton3 supercomputer. Additionally, CholBindNet achieved strong model interpretability through atomic-level feature encoding, Grad-CAM visualization, and attention-based scoring analysis. Overall, CholBindNet provides an efficient and scalable approach to classify and rank cholesterol binding sites on membrane proteins, achieving performance comparable to computationally expensive MD simulations while providing rich biophysical insights into atomic-level spatial patterns beyond amino acid sequences. This study lays the foundation for future deep learning models targeting drug binding sites in membrane proteins and cholesterol-modulating therapeutics.


