image:
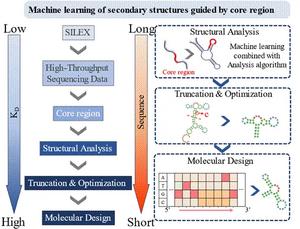
Figure 1. Flowchart of the working principle of machine learning based on single-round nucleic acid aptamer screening sequences. This includes deep learning-driven molecular design based on deep learning of core sequences, machine learning analysis of secondary structure, truncation and optimization processes, and single-round screening sequences.
view more
Credit: CCS Chemistry
A research team led by Weihong Tan, Xiaohong Fang, and Tao Bing from the Hangzhou Institute of Medical Sciences, Chinese Academy of Sciences, has proposed a new method for analyzing nucleic acid aptamer sequences based on machine learning. This method can directly analyze the secondary structure of nucleic acid aptamers from one round of screening data, so detailed secondary structure information of nucleic acid aptamers can be obtained without iterative enrichment. This enables rational cleavage and optimization of high-affinity nucleic acid aptamers as well as the design of nucleic acid aptamer molecules, greatly accelerating the process of nucleic acid aptamer discovery and optimization. This paper is an open access research paper. CCS Chemistrythe flagship journal of the Chinese Chemical Society.
Background information:
Nucleic acid aptamers are nucleic acid molecules that specifically recognize target molecules, and their secondary structures are highly diverse and exhibit complex three-dimensional structures. Although SELEX technology generates a large number of candidate sequences, determining the functional secondary structure for binding to the target is difficult. Furthermore, the nucleic acid aptamers obtained by screening may not be the optimal nucleic acid sequences and require further optimization. However, traditional structural characterization techniques such as electron microscopy, nuclear magnetic resonance, and X-ray crystallography are insufficient to efficiently resolve their structures, severely limiting the cleavage optimization and molecular design of nucleic acid aptamers.
Highlights of this article:
To address these issues, the team established a machine learning-based analysis method (shape 1). This method utilizes unsupervised autoencoder clustering and deep learning to analyze core sequences within an aptamer family in a single screening. These core sequences are then used as an index to analyze a large amount of secondary structure using machine learning strategies to extract secondary structure features common to nucleic acid aptamers. This strategy not only achieves rational cleavage and performance optimization of nucleic acid aptamer sequences, but also provides a new approach for de novo design and generation of specific nucleic acid aptamer sequences.
To obtain common core sequences of nucleic acid aptamers from single-round screening libraries, the authors used deep learning to systematically analyze family distribution patterns in single-round aptamer screening sequences of CD8 protein (shape 2). The results showed that although the sequence background was highly heterogeneous, most families were enriched with the same core sequence ‘GTGAGGAGCTTGAAA’. Conventional multiple sequence alignment methods cannot effectively extract this important homologous sequence information because it is difficult to handle short motifs with low homology background.
To verify the accuracy of the core sequence obtained in the screening, the authors synthesized a library containing a portion of the core sequence (5′-AGCTTGAAA-3′) and performed RE-SILEX. All of the more than 20,000 nucleic acids obtained from the screen contained core sequences that matched those obtained from a single screen. To clearly elucidate the structure-function relationship between the aptamer and its target, a machine learning-based algorithm was further developed to analyze the secondary structure mediated by the aptamer core sequence in a single screening. For the fixed region sequence “5′-AGCTTGAAA-3′”, 62.4% of the sequence (total 24,867) formed stem-loop secondary structures, while the remaining 37.6% aptamers were able to form other secondary structures within the fixed region. Among aptamers that form stem-loop structures in fixed base regions, the sequence “GTGA” appeared in 55.2% (13,711 copies) of multibranched loops and 44.8% (11,301 copies) of stem structures (shape 3c). The authors quantified the length of individual stem loops within the hyperbranched loop and analyzed the base distribution at each position. Through deep mining of the secondary structure of different sequences using machine learning, they reasoned that these sequences form a covalent secondary structure that can bind to the same epitope of the target (shape 3D). Based on this secondary structure, the authors cut and optimized the nucleic acid sequence obtained from RE-SILEX, significantly increasing its affinity. This means that we have successfully obtained over 10,000 potentially active CD8-specific aptamers.
To further confirm the applicability of this method to the analysis of single-round aptamer screening data, the authors used this method to analyze CD8 single-round aptamer data. Of the first 1,000 sequences of single-round aptamers for the CD8 protein, 770 sequences contained a core sequence, and the majority of sequences exhibited highly consistent shared secondary structure characteristics (shape 4). Based on the shared secondary structure properties of the CD8 protein, these aptamers were cleaved and optimized to successfully increase affinity by an order of magnitude while maintaining high specificity and accurate recognition of the CD8 protein even in complex cellular microenvironments. Furthermore, based on the secondary structure features of the CD8 protein aptamer, it not only allows the design of split-type aptamers, but also opens up new possibilities for de novo aptamer design.
To verify the universality of the aforementioned machine learning analysis method, the authors further applied it to single-round screening data analysis of fibroblast activation protein (FAP). The results show that the FAP aptamer family is also enriched in a highly conserved core sequence (5′-GGGGTCTGCTTCGGATTGCGG-3′), suggesting that the core sequence forms a G-quadruplex structure, while its two terminal sequences can form a hairpin structure to stabilize the G-quadruplex structure (shape 5). Based on this common secondary structure, FAP protein aptamers were truncated and optimized to significantly improve binding affinity. This successful application fully demonstrates the universality of the machine learning-enabled single-round aptamer analysis method when dealing with aptamers of different structural types.
Overview and outlook:
In summary, this study shows that single-round nucleic acid aptamer screening libraries already contain rich structural information that was previously thought to require multiple rounds of screening. By combining high-throughput sequencing and machine learning, the authors developed a method that can decipher the secondary structure of nucleic acid aptamers and pinpoint targets that bind to key conserved functional motifs. This technology achieves rational cleavage and optimization of nucleic acid aptamers, increasing affinity by more than 10-fold without resorting to tedious experimental structural analysis. This study not only improves the efficiency of nucleic acid aptamer discovery but also challenges the traditional paradigm by highlighting the dominant role of spatial structure in molecular recognition. This research approach also opens new avenues for designing functional nucleic acids, exploring non-coding RNA-protein interactions, and developing AI-driven virtual screening platforms for nucleic acid aptamers, driving the rapid development of next-generation nucleic acid aptamer technologies for precision diagnosis and therapy.
This research was supported by the National Natural Science Foundation of China, the Zhejiang Provincial “Pioneer” and “Leading Goose” Research and Development Programs, the Zhejiang Provincial Natural Science Foundation, the National Health Commission Scientific Research Fund-Zhejiang Provincial Health and Welfare Key Science and Technology Program, and the Strategic Priority Research Program of the Chinese Academy of Sciences.
—
About the diary: CCS Chemistry is the flagship publication of the Chinese Chemical Society and was established to serve as the premier international chemistry journal published in China. This is an English journal covering all areas of chemistry and chemical science, including breakthrough concepts, mechanisms, methods, materials, reactions, and applications. All articles are Diamond Open Access, meaning there are no fees for either authors or readers. For more information, please visit https://www.chinesechemsoc.org/journal/ccschem.
About the Chinese Chemical Society: The Chinese Chemical Society (CCS) is an academic organization voluntarily established by Chinese chemists with the aim of uniting Chinese chemists at home and abroad and promoting the development of chemistry in China. CCS was founded on August 4, 1932 during a conference of prominent chemists held in Nanjing. We currently have over 120,000 individual members and 184 institutional members. There are seven divisions covering the major areas of chemistry: physical, inorganic, organic, polymer, analytical, applied, and chemical education, and 31 committees, including catalysis, computational chemistry, photochemistry, electrochemistry, organic solid state chemistry, environmental chemistry, and many other subfields of chemical science. CCS also has 10 committees, including the Women Chemists Committee and the Young Chemists Committee. For more information, please visit https://www.chinesechemsoc.org/.
Research method
Computational simulation/modeling
Research theme
not applicable
Article title
Single-round aptamer discovery using machine learning: elucidating the structure-function principle of target binding
Article publication date
January 7, 2026
Conflict of interest statement
There are no conflicts of interest to report.
Disclaimer: AAAS and EurekAlert! We are not responsible for the accuracy of news releases posted on EurekAlert! Use of Information by Contributing Institutions or via the EurekAlert System.