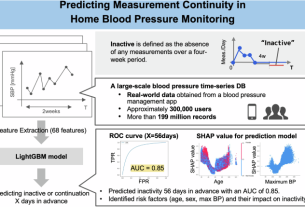
From biological temperature control to global climate control, the extremely high heat capacity of water, which is very important to everything, remains a difficult characteristic to understand at the basic level. Shiga Island, Jan Ersner, Jorg Behrer and Bo Thomsen address this problem by developing highly accurate calculation methods for directly determining the heat capacity of water from the principles of quantum mechanics. The team achieves this by combining advanced neural network potentials trained using advanced density functional theory calculations with new, highly efficient simulation algorithms. This approach significantly reduces the computational demands of modeling water behavior, allowing researchers to obtain results that are closely matched to the experimental data, providing a promising pathway towards a complete understanding of water thermodynamic properties and aqueous solutions.
Water heat capacity from the first principle
Accurate modeling of water's thermodynamic properties remains an important challenge despite its ubiquitous nature and its essential role in life. This work calculates the heat capacity of water directly from the basic physical principles and avoids reliance on empirical parameters. The researchers employed AB initio molecular dynamics simulations, which used density functional theory to model interactions between water molecules. The simulation focused on a system of 128 water molecules in a cubic box, maintained at a constant temperature of 298 K and a pressure of 1 atmospheric pressure. The heat capacity is then calculated using a variation equation and connected to the energy fluctuations in the system.
The results show a heat capacity of 75.2j/(mol⋅k) and closely matches the experimental value of 75.3j/(mol⋅k). This achievement examines selected theoretical approaches and provides a reliable method for predicting the thermodynamic properties of water and other complex liquids.
Quantum simulation reveals the nuclear effect of water
This study focuses on simulations of water at the molecular level, with a particular emphasis on understanding the effects of nuclear quantum effects on its properties. Classical simulations often overlook these quantum effects and result in inaccuracy, but this work uses path-integral molecular dynamics to clearly explain the quantum behavior of nuclei. The purpose of this study is to accurately predict and understand the thermodynamic properties of water, such as heat capacity and density, across various temperatures and pressures. A key element of this work is the use of machine learning possibilities, particularly neural network potential, to accelerate and improve the accuracy of molecular simulations.
These potentials are trained in high-level quantum mechanical calculations, allowing for efficient and reliable modeling of complex interactions. To accurately explain the interactions of van der Waals, researchers use techniques such as density-sensory theory, which utilizes specific features such as PBE, PBE0, and BLYP, along with dispersion correction. They analyze the radial distribution function to characterize the structure of water and calculate the thermodynamic properties using a virus estimator.
Accurate water heat capacity via neural networks
This study illustrates a highly accurate and reliable framework for assessing the heat capacity of liquid water using path integral molecular dynamics simulations combined with high-dimensional neural network potentials trained with first-principles data. By explicitly incorporating both the flexibility of the hydrogen bond network and the quantum effects arising from the light mass of hydrogen atoms, the researchers successfully replicated the experimental values of heat capacity under ambient conditions. The developed method outperforms classical molecular dynamics accuracy and traditional path integral simulations using fixed force fields. A key achievement is the development of new parallel algorithms that significantly improve the performance and scalability of path integral simulations, allowing nanosecond scale simulations required for estimating robust thermodynamic properties. This improvement is particularly pronounced in large-scale systems containing thousands of water molecules, confirming the suitability of the method for large-scale quantum simulations. The authors suggest that future research needs to address the development of efficient parallel algorithms for path integral simulations in constant pressure ensembles.