image:
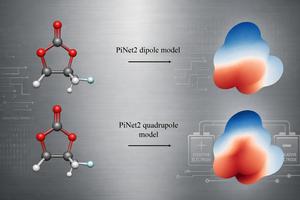
Molecular electrostatic potential of fluoropropylene carbonate (FPC) generated from PiNet2 model with physical constraints
view more
Credit: Kadri Muuga and Chao Zhang, Uppsala University
Electrolytes are at the heart of modern electrochemical energy storage. These control how ions move, how interfaces form, how stable the battery is over time, and ultimately how safe and efficient the device is. However, discovering better electrolyte molecules remains a very difficult problem. The associated behavior depends on subtle intermolecular interactions, solvation effects, and charge distributions, which are often expensive to resolve using quantum chemical calculations at the scale required for materials discovery. This is where artificial intelligence (AI) is starting to stir things up. By learning from quantum data, machine learning (ML) models can help explore chemical space much faster than traditional electronic structure workflows.
The important quantity in this problem is the molecular electrostatic potential (MEP). MEP maps how molecules experience electrostatic attraction and repulsion in the space around them and is widely used to understand intermolecular interactions, molecular recognition, reactivity, and solvent design. However, in actual simulations, the complete continuous electrostatic potential is rarely handled directly. This is because obtaining the electrostatic potential accurately is computationally intensive and takes days or even weeks to calculate.
In a new study, a research team from Uppsala University investigated whether ML models can efficiently infer MEPs from molecular multipole information. The researchers used the PiNet2 architecture to train the model on dipole and quadrupole moments and tested it on the widely used QM9 dataset, a simple dataset of organic molecules ideal for studying fundamental chemical properties, and the broader and more complex SPICE dataset. They found that including the quadrupole moment significantly increases the model’s ability to reconstruct the electrostatic potential compared to a model that only includes dipoles. The same trends were found in both datasets.
This study shows that ML models trained on quadrupole moments enable rapid and accurate prediction of MEPs, providing an efficient alternative to computationally intensive quantum computations. This approach supports high-throughput screening and solvent design of energy storage devices by facilitating the characterization of electrostatic interactions in electrolytes and solvent molecules. The results further demonstrate that quadrupole moments constitute a more effective training target than dipole moments in ML-based charge models and provide a practical framework for accessing electrostatic information. Overall, this methodology can accelerate the discovery and optimization of safer, stable, and high-performance battery solvents.
The study also highlights broader lessons for AI in chemistry. This means that the choice of training target is very important. Dipoles are the key term in the multipole expansion of MEPs for neutral molecules and are therefore often treated as natural first targets. However, this study shows that quadrupoles carry particularly valuable information when the goal is to recover the electrostatic landscape from simple point charges.
The research was carried out at Uppsala University and was supported by the European Research Council, the Wallenberg Initiative Materials Science for Sustainability and the Swedish Energy Agency.
References: Kadri Muuga, Lisanne Knijff, Chao Zhang. Molecular electrostatic potentials from machine learning models for dipole and quadrupole prediction[J]. AI for science. DOI: 10.1088/3050-287X/ae531a
Article title
Molecular electrostatic potentials from machine learning models for dipole and quadrupole prediction
Article publication date
March 17, 2026
Disclaimer: AAAS and EurekAlert! We are not responsible for the accuracy of news releases posted on EurekAlert! Use of Information by Contributing Institutions or via the EurekAlert System.

